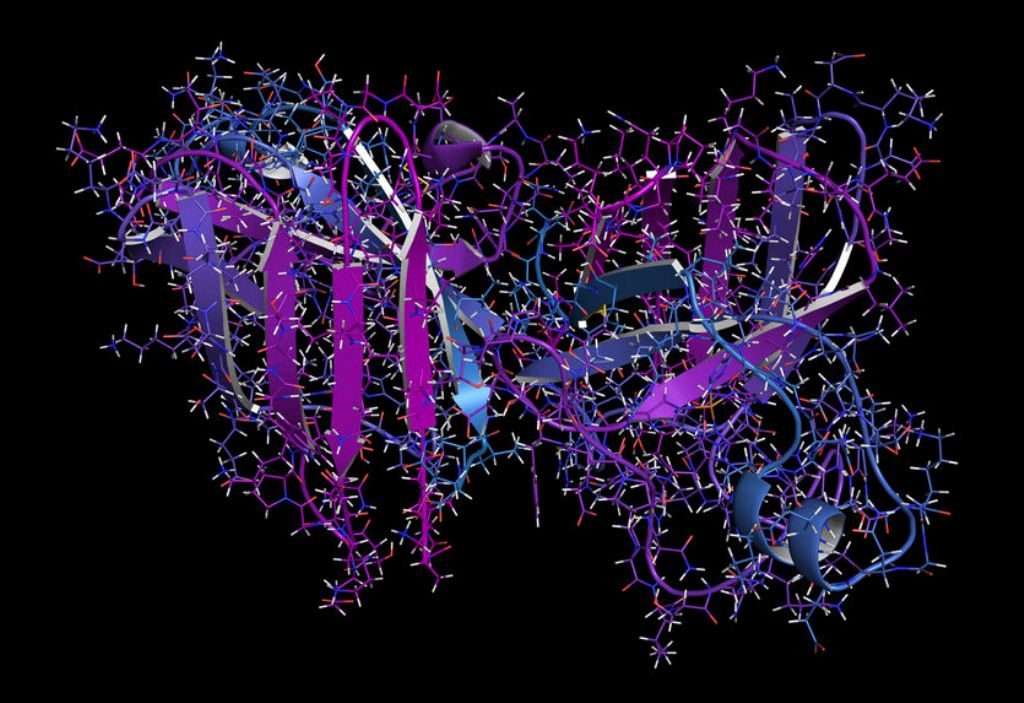
40894232 – superoxide dismutase 1 (sod1) enzyme. converts superoxide radical in hydrogen peroxide. gene mutations cause als (amyotrophic lateral sclerosis). cartoon + line representation. n-to-c gradient coloring.
HONGOS Y ELA
La esclerosis lateral amiotrófica o ELA, es una enfermedad de las neuronas en el cerebro, el tronco cerebral y la médula espinal que controlan el movimiento de los músculos voluntarios.
La ELA también es conocida como la enfermedad de Lou Gehrig.
LA ELA ES UNA DE LAS ENFERMEDADES MÁS TERRIBLES QUE CONOCEMOS
La enfermedad suele atacar entre los 40 y los 60 años y es más común entre los hombres que entre las mujeres. No se conoce la causa de la enfermedad. Puede ser parte de una tendencia familiar, pero generalmente se presenta aleatoriamente. No existe una cura. Las medicinas pueden aliviar los síntomas y, algunas veces, prolongar la supervivencia.
Síntomas
Los signos y síntomas de la ELA varían mucho de una persona a otra, según qué neuronas estén afectadas. Algunos signos y síntomas son:
Dificultad para caminar o realizar actividades diarias normales
Tropezones y caídas
Debilidad en las piernas, los pies o los tobillos
Debilidad o torpeza en las manos
Dificultad para hablar o problemas para tragar
Calambres musculares y espasmos en brazos, hombros y lengua
Llanto, risa o bostezos inapropiados
Cambios cognitivos y de comportamiento
La ELA con frecuencia comienza en las manos, los pies o las extremidades y luego se extiende a otras partes del cuerpo. A medida que la enfermedad avanza y las células nerviosas se destruyen, los músculos se debilitan. Esto eventualmente afecta la masticación, la deglución, el habla y la respiración.
Generalmente no hay dolor en los estadios tempranos de la ELA, y el dolor es poco común en los estadios avanzados. La ELA no suele afectar al control de la vejiga ni a los sentidos.
Causas
La ELA afecta las células nerviosas que controlan los movimientos voluntarios de los músculos, como caminar y hablar (neuronas motoras). La ELA hace que las neuronas motoras se deterioren gradualmente y luego mueran. Las neuronas motoras se extienden desde el cerebro hasta la médula espinal y los músculos de todo el cuerpo. Cuando las neuronas motoras están dañadas, dejan de enviar mensajes a los músculos, por lo que los músculos no pueden funcionar.
La ELA se hereda en el 5 % al 10 % de las personas. Se desconoce la causa en el resto de las personas.
Los investigadores continúan estudiando las posibles causas de la ELA. La mayoría de las teorías se centran en una interacción compleja entre los factores genéticos y ambientales.
Factores de riesgo
Estos son algunos de los factores de riesgo establecidos para la esclerosis lateral amiotrófica:
Factor hereditario. Entre el 5 y el 10 % de las personas con esclerosis lateral amiotrófica la heredaron (esclerosis lateral amiotrófica familiar). La mayoría de los hijos de personas con esclerosis lateral amiotrófica familiar tienen un 50 % de probabilidades de desarrollar la enfermedad.
La edad. El riesgo de esclerosis lateral amiotrófica aumenta con la edad, y es más común entre los 40 y mediados de los 60 años.
Sexo. Antes de los 65 años, la esclerosis lateral amiotrófica es un poco más común en hombres que en mujeres. Esta diferencia de sexo desaparece después de los 70 años.
La genética. Algunos estudios que examinan todo el genoma humano encontraron muchas similitudes en las variaciones genéticas de las personas con esclerosis lateral amiotrófica familiar y algunas personas con esclerosis lateral amiotrófica no hereditaria. Estas variaciones genéticas podrían hacer que las personas sean más susceptibles a la esclerosis lateral amiotrófica.
Los siguientes factores ambientales podrían desencadenar la esclerosis lateral amiotrófica.
Tabaquismo. Fumar es el único factor de riesgo ambiental probable de la esclerosis lateral amiotrófica. El riesgo parece ser mayor para las mujeres, particularmente después de la menopausia.
Exposición a toxinas ambientales. Cierta evidencia sugiere que la exposición al plomo u otras sustancias en el lugar de trabajo o el hogar podría estar relacionada con la esclerosis lateral amiotrófica. Se han realizado muchos estudios, pero ningún agente o sustancia química ha sido asociado consistentemente con la esclerosis lateral amiotrófica.
Servicio militar. Los estudios indican que las personas que han servido en las Fuerzas Armadas tienen un mayor riesgo de sufrir de esclerosis lateral amiotrófica. No está claro qué podría desencadenar el desarrollo de esclerosis lateral amiotrófica. Podría incluir la exposición a ciertos metales o productos químicos, lesiones traumáticas, infecciones virales y esfuerzo intenso.
Es una patología neurodegenerativa y progresivo, con una múltiple cantidad de mutaciones en diversos genes, que tienen como resultado su principal manifestación clínica que es la afectación de la neurona motora superior (primera motoneurona), y neurona motora inferior (segunda motoneurona), siendo este el fenotipo de la forma clásica presente en un 80 % de los casos. Existen otras variantes fenotípicas donde predomina específicamente un componente clínico las cuales son: Esclerosis lateral primaria (afectación neurona motora superior), Atrofia muscular progresiva (afectación de neurona motora inferior), y parálisis bulbar progresiva (sintomatología bulbar). Es necesario aclarar que no es una patología puramente motora ya que también existen afecciones extra motoras como alteraciones cognitivas y del comportamiento.
Una media de vida 3 a 5 años
La esclerosis lateral amiotrófica tiene una supervivencia realmente corta de 3 a 5 años en su forma clásica (mayor edad de inicio entre 65 y75 años), y en sus otras variantes llega a alcanzarse 20 años o más, encontrando que es menos letal cuanto más temprano sea la edad de inicio. Predomina con mayor frecuencia en hombres.
Respecto a su etiología, si bien es cierto que puede ser causada por diversas mutaciones genéticas las más comunes son: Mutación del gen C9orf72 en 40 % de las formas familiares y 8 % de las esporádicas; Mutación del gen SOD1(participa en mecanismos de defensa celular contra radicales libres) en 10-20% de las formas familiares y esporádicas. Por otro lado, se investigan aun alguna posible asociación con factores de riesgo ambientales o tóxicos.
Clínicamente se presenta con un déficit motor por paresia y atrofia muscular principalmente en las extremidades de carácter simétrico y que progresa rápidamente teniendo como desenlace el fallo respiratorio. Algunas otras manifestaciones con menor frecuencia, que son características de afección bulbar son disfagia y disartria. Es muy importante decir que no hay afectación motora en la musculatura ocular ni alteraciones de tipo sensitivo lo cual es un gran refuerzo para el diagnóstico diferencial de otras entidades patológicas similares. Recordemos que el predominio de algunas manifestaciones clínicas nos ayudara a definir a alguna variante de la esclerosis lateral amiotrófica.
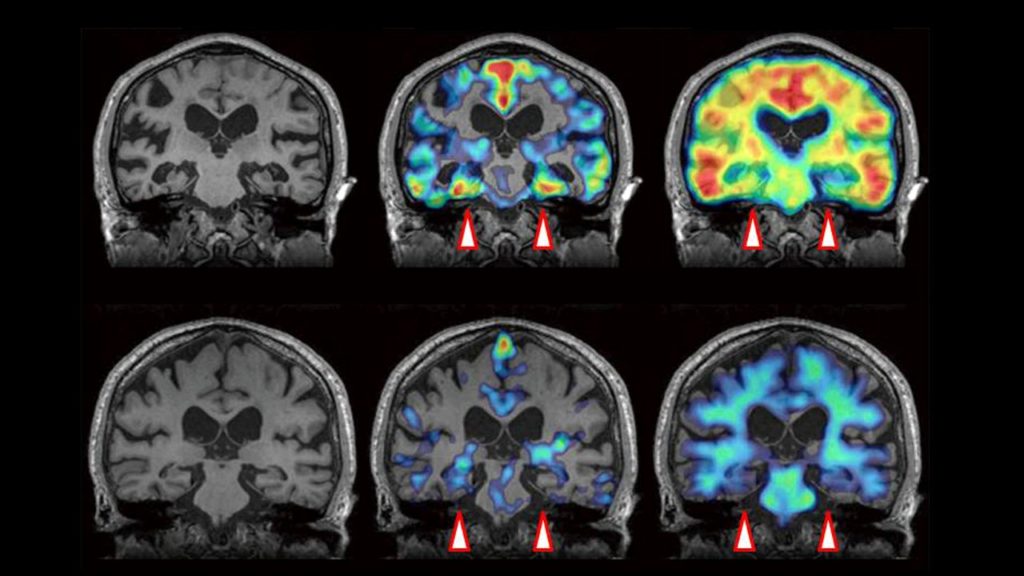
La fisiopatología del alzhéimer y la ELA, clave para adelantar el diagnóstico
La esclerosis lateral amiotrófica (ELA), el párkinson y el alzhéimer afectan a la capacidad cognitiva y están cada vez más presentes en la sociedad. No tienen cura y, pese a que se han dado pasos importantes, aún no se conocen bien sus causas, por eso estudiar su fisiopatología sigue siendo la clave.
Una serie de personalidades científicas han sufrido esta patología de una manera dramática que pasó a describir
La ELA
El científico británico Stephen Hawking sufrió esclerosis lateral amiotrófica. EFE/TAL COHEN
Jeffrey D. Rothstein, director del Brain Science Institute (Baltimore, EE.UU.), es experto en ELA, una enfermedad con una incidencia anual de tres casos por cada 100.000 habitantes en España.
Afecta a las neuronas motoras de la corteza cerebral, al tronco cerebral y a la médula espinal y produce una debilidad y atrofia muscular progresiva. Sus causas aún no se conocen pero en “los últimos años se han producido descubrimientos clave”, asociando alteraciones en el transporte de proteínas a nivel celular con ésta.
“Los avances en la genética de la ELA suponen para nosotros una revolución”, señaló este investigador, quien insiste en que entender sus mecanismos es lo que abre la puerta a los tratamientos.
En cuanto a si en este tipo de enfermedades se invierten menos recursos, este científico ha remachado que “todo el mundo pide más dinero”, aunque reconoció que es una “ecuación complicada”.
A su juicio, es una cuestión de retorno, pero en el caso de los pacientes con enfermedades neurodegenerativas el gasto para las arcas públicas es altísimo, lo que habría que tener en cuenta.
Para Arthur Konnerth, del Instituto de Neurociencia de la Universidad Técnica de Múnich (Alemania) e investigador en la enfermedad de Alzheimer, no es tanto la inversión, en este caso de la industria, sino que se pueda investigar con primates, que “sería fundamental”.
El alzhéimer
El envejecimiento activo busca la participación social de las personas mayores. Este concepto es cada vez más visible en la sociedad. EFE/Kiyoshi Ota
Kommerth explicó que el alzhéimer es la principal causa del declive intelectual en la población mundial ya que se caracteriza por la pérdida progresiva de memoria, funciones cognitivas y demencia.
En la actualidad se conoce aún muy poco sobre las causas que llevan al deterioro de las funciones cerebrales en la enfermedad, pero una característica esencial es la acumulación de proteína beta-amiloide en el cerebro, ha subrayado este experto.
Koomerth, quien señaló que otra de las cosas implicadas en alzhéimer es el mal funcionamiento de los circuitos cerebrales, detalló que muchos ensayos con fármacos no terminan de funcionar -el 60 % de los ensayos clínicos en fase II se abandonan- porque aún no entienden a fondo las causas tempranas de la enfermedad y porque se trata de patologías de progreso lento: “no sabemos cuándo empiezan”.
Christian Haass, jefe de la división de Bioquímica de la Universidad Ludwig-Maximilians (Múnich), dijo que parte de la esencia de las investigaciones ahora está en tratar de prevenir la producción del péptido beta-amiloide: un “abordaje precoz”.
Hace unos días vuelve a aparecer uno de los varios trabajos del trabajos publicados por el Doctor Carrasco, de la facultad de medicina de Madrid
He visto publicados varios trabajos de este autor en los que afirma de una manera muy da que se etiología está claramente relacionada con los hongos , y lo repite y parece tan lógico que llama la atención que otros autores no corroboren estos hallazgos.
Recientemente he tenido ocasión de leer una publicación
La presencia de hongos en pacientes con ELA nos vuelve a ilusionar con el origen microbiano de la enfermedad. Estas aéreo clínico lleva ya varios años en los medios y de difícil de entender cómo sólo el Dr. Carrasco, habla sólo del tema
–
Crédito de
El grupo de investigación que lidera Luis Carrasco, catedrático de microbiología de la Universidad Autónoma de Madrid (UAM), ha vuelto a detectar hongos en el sistema nervioso central de pacientes fallecidos con esclerosis lateral amiotrófica (ELA).
El profesor Carrasco y sus colegas sostienen desde hace años que la ELA y otras enfermedades neurodegenerativas como el alzhéimer tienen un origen microbiano, específicamente en infecciones fúngicas que, dependiendo de la enfermedad, varía la vía de infección, las regiones infectadas y el tipo de especies.
En una entrevista que realizamos a Carrasco en 2015 en relación a los descubrimientos de hongos en pacientes fallecidos con alzhéimer, opinó que la vía más directa para saber si sus hipótesis —cuestionadas por muchos— son o no acertadas, es hacer un ensayo clínico con fármacos antifúngicos. La ELA podría estar causada por hongos en el cerebro
Hasta diez veces he visto publicadas y siempre por el Dr. Carrasco la relación entre hongos y
ELA, pero no la he vuelto a ver publicada por nadie más parece muy lógica y sobre todo muy apetecible.
Sigo teniendo presente: el algoritmo
La alteración de la microbiota
Desequilibró de los gérmenes que la componen
En migración de los mismos
Alineación en regiones varios de nuestra economía
La invasión de esta región es por macrófagos y la enfermedad neurodegenerativa
Está claro que esto no debe ser así, no hay publicaciones sobre el tema. Recientemente vuelvo a encontrar una publicación sobre el tema
¿Hongos? Una noticia que nos ha cogido a absolutamente todos con el pie cambiado
Un grupo de científicos españoles ha publicado un estudio que apunta que una compleja infección por hongos podría ser clave para desarrollar esclerosis lateral amiotrófica
CARLOS MATALLANAS
Este es el artículo número 32 del presente blog y, sin duda, hasta ahora no he afrontado ninguno tan complejo de escribir. Seguro que toda la comunidad de pacientes de ELA, nuestros seres queridos y los profesionales que nos atienden estarán al corriente de la noticia. Para el resto, la recuerdo rápidamente en este inicio.
Un grupo de científicos españoles publicó el último día de abril un pequeño estudio pero cuyas conclusiones son, en sí mismas, muy sorprendentes. Según sus resultados, una compleja infección por hongos podría ser clave para desarrollar esclerosis lateral amiotrófica.
Estos profesionales del Centro de Biología Molecular Severo Ochoa, sustentado por la Universidad Autónoma de Madrid (UAM) y el Centro Superior de Investigaciones Científicas (CSIC), analizaron muestras de tejido cerebral y de líquido cefalorraquídeo de fallecidos por ELA y encontraron varias especies de hongos, en abundancia y de forma diseminada. Elementos no hallados en los cadáveres sin ELA usados para el experimento.
Según Luis Carrasco, catedrático de Microbiología de la UAM y responsable del estudio, el sistema que ellos han elaborado para la detección de estos hongos no deja lugar a dudas, por lo que entiende que los resultados son rotundos y esclarecedores. Si bien, con ellos simplemente han podido elaborar una hipótesis del posible origen de la ELA, instando al resto de la comunidad científica a seguir esa teoría para comprobarla, completarla y darle validez y uso, llegado el caso.
El estudio del doctor Carrasco y su equipo lanza básicamente una hipótesis, que debe ser confirmada primero, y completada a continuación
La rotundidad de esas conclusiones, publicadas en una modesta revista científica (International Journal of Biological Sciences), hizo que la prensa generalista lanzara rápidamente la noticia a la sociedad. El mismo jueves, pasados unos pocos minutos de la una de la tarde, escuché en el boletín informativo de la emisora de radio que tenía puesta un titular espectacular: “Hallan la causa de la esclerosis lateral amiotrófica”. La frase, digna de los mejores dramas de Hollywood, me hizo dar un respingo y subir el volumen para no perder detalle al desarrollo de la información. Ahí todo se atenuó bastante al escuchar al propio Luis Carrasco dando, a grandes rasgos, las nociones que les acabo de explicar líneas más arriba.
De una a dos, a Marta y a mí nos dio tiempo a buscar bien la publicación científica, leerla con detalle, mandársela a la gente que conocemos que saben bien sobre todo esto, calmar a los familiares y amigos que seguro al enterarse podrían estar viviendo o iban a vivir un momento de euforia aún injustificado y, por supuesto, dentro de la mesura, también nos dio tiempo para emocionarnos.
Cuando dieron las dos, la misma emisora seguía incluyendo la noticia en el boletín, si bien (seguro que alertados por lo que significa no ser fieles a la estricta realidad en algo tan delicado), el titular fue reescrito y redactado sin tanta rotundidad. Algo así como “científicos españoles hallan una posible causa de la esclerosis lateral amiotrófica en un estudio preliminar”. Habrá quien piense que ambas afirmaciones son prácticamente la misma, pero tanto por mi condición de periodista como por la de afectado por la noticia, les aseguro que son como la noche y el día. La primera es casi temeraria, la segunda es precisa e informativamente correcta.
Esa misma tarde, a mi hermano Javier le llamaron del programa La Ventana de la Cadena Ser para entrar en directo y hablar acerca del hallazgo. Habló después del doctor Carrasco, que explicó más profundamente su estudio, y mi hermano simplemente dio nuestra visión particular de como una familia afectada por la ELA había reaccionado ante esto.
[‘Cryptococcus spp’. y ‘Malasezzia spp’ y ‘Candida albicans’, los tres hongos que podrían estar relacionados con la ELA. (Creative Commons)]
‘Cryptococcus spp’. y ‘Malasezzia spp’ y ‘Candida albicans’, los tres hongos que podrían estar relacionados con la ELA. (Creative Commons)
Una llamada a la calma
Explicado todo el mapa de la novedad en sí, ahora intentaré ser lo más responsable posible, como me obliga el seguimiento que tiene este blog. Como a los periodistas que se lanzaron a la piscina para luego rebajar sus eufóricas palabras, la publicación del estudio también ha cogido con el pie cambiado a todas las personas implicadas. En las redes sociales, a lo largo del puente, se fueron enviando de aquí para allá las noticias de los distintos medios que recogían el resultado del estudio. Así que supongo que la expectación que sentimos en mi familia será compartida por todas las que están en la misma situación.
Es analizando todo el fenómeno en su conjunto donde toca llamar a la calma y a la prudencia. Es fenomenal que se siga investigando, y cada vez se hace más y con más recursos. Solo así se ganará a la ELA y la humanidad logrará dominar a esta asesina y a otras que se le parecen, aunque no sean tan crueles y devastadoras. Pero el estudio del doctor Carrasco y su equipo lanza básicamente una hipótesis, que debe ser confirmada primero, y completada a continuación. Quizá con demasiada ligereza, el propio investigador pedía a los enfermos en una entrevista publicada este lunes que instasen a los respectivos médicos a buscarles hongos y a tratarlos de inmediato si se encontraban. Y esta iniciativa creo que debe tenerla siempre el facultativo, jamás el enfermo. Si bien, no está de más que cualquier enfermo informe sobre este estudio a su médico, por si no está al corriente de ello. Y será este quien deba tomar cualquier decisión al respecto o recomendarle cualquier línea a seguir si lo ve necesario.
Si los enfermos debemos tener el talante de aguantar y no dejar que la euforia nos embriague, los científicos están obligados a mantenerse expectantes
Esos son los tiempos de la ciencia, y hay que respetarlos. Ya sabemos que la enfermedad avanza a una velocidad mayor y da dolor a quienes la sufren no poder encontrar nunca algo a lo que agarrarse para mejorar. O al menos, algo con lo que dejar de empeorar día a día. Pero aquí no hay elección, cada uno debe permanecer en su trinchera, peleando en su día a día contra todas las bombas que le caen sin cesar y haciendo todo lo posible por mantenerse lo más entero posible. Llegará un momento, seguro, que los enfermos de ELA empezarán a salir de esa trinchera. Y será la gente de ciencia quien encuentre el camino seguro. Lógicamente, hasta que eso llegue, seguirán cayendo muchos dentro de estas crueles trincheras donde estamos confinados. Es imposible saber a día de hoy si cada uno de nosotros será uno de los elegidos o las ayudas llegarán demasiado tarde en nuestro caso concreto.
Eso por el lado de los enfermos. Pero la misma mesura, aunque en sentido contrario, le pido modestamente desde aquí a los científicos y profesionales. Nos consta que hay expertos en la investigación de la ELA que han acogido con demasiado escepticismo los resultados del estudio. Está claro que tiene elementos muy mejorables, empezando por el número de muestras (solo se analizaron tres cadáveres con la enfermedad y cinco sin ella) y pasando por la falta de explicación de algunos pormenores del sistema de análisis que hacen dudar a los más puristas.
Aun así, es totalmente imprudente y nada inteligente rechazar de plano los resultados. Si los enfermos debemos tener el talante de aguantar y no dejar que la euforia nos embriague, los científicos están obligados (por ética profesional y respeto profundo a la condición humana) a mantenerse expectantes ante lo expuesto por este estudio de investigadores españoles. Y por supuesto que los más críticos (que ya los ha habido a nivel internacional) deberían ser los primeros en intentar comprobar si de verdad hubo fallos o si los resultados no son del todo tan rotundos o convincentes.
Para ello, si he entendido bien a los expertos que me han informado, lo primero es repetir el estudio de manera más completa en un ambiente y escenario diferente. Si todo sale igual, habrá motivos firmes para la alegría y lo siguiente será empezar a desarrollar ensayos con pacientes, donde entra la medicina y las farmacéuticas. Según la hipótesis del doctor Carrasco, este paso sería mucho más sencillo que habitualmente, al ser las infecciones fúngicas bastante conocidas y existir tratamientos efectivos desde hace años y de uso común en la sanidad mundial. Lo que no quita que hubiara que averiguar el correcto reajuste de los mismos para tratar algo que se intuye lo suficientemente complejo como para llevar decenas de años escondido del saber médico.
Paralelamente, debería continuar también la búsqueda de respuestas, acerca de cómo se daría esa infección, si los hongos son causa o consecuencia del proceso, como influyen exactamente en el fallo de las motoneuronas, qué tipo de genética tienen las personas a las cuales les apareció la enfermedad, y un largo e indeterminado etcétera. Porque no se trata solo de salvar a los enfermos actuales. Dominar una enfermedad significa que se pueda prever su aparición, tratarla una vez aparecida y, por supuesto, el fin último, curarla. Por eso, y lo recuerdo de nuevo, estos avances nos incumben a todos.
Se pide celeridad, eficiencia y eficacia
En definitiva, la pelota está en el tejado de los expertos, institutos y centros investigadores de la ELA, es ahí donde corresponde demostrar que Carrasco está equivocado o que acierta. Entiendo que les haya cogido a ellos también con el pie cambiado, como a todos, pero se pide celeridad, eficiencia y eficacia científica. Y creo que la espectacularidad de lo hallado merece ser confirmado o descartado lo antes posible. Las creencias, las suposiciones y la suficiencia profesional deben permanecer fuera de los laboratorios. La gente de la calle no necesita ese rigor para subsistir, pero el científico sí, y sin él es inviable que haga bien su trabajo. Yo soy ajeno a ese mundo, pero el propio sentido común me hace pensar de esa manera.
Les he comentado alguna vez que los hallazgos de un pequeño laboratorio de un rincón del planeta donde apenas hay recursos pueden resultar clave para encontrar respuestas importantes que ni en los centros más especializados y con más recursos lograron dar con ellas. Es la grandeza del saber y la ciencia, una suma de trabajos continua que no cesa. Podríamos estar ante uno de esos casos, de esas carambolas inesperadas que marcan un hito. O no, podría resultar que las muestras estaban contaminadas o el sistema fue erróneo o algo desvirtuó los resultados. Profesionales, como son, de nuestras universidades y colaboradores del CSIC sabemos que tienen una preparación de primer orden mundial. Pero como en todo, aquí también hay egos, y este pequeño estudio no está dentro de las líneas generales de investigación mundial de la ELA. De ahí supongo el escepticismo que ha levantado de primeras.
A todos nos gustaría que ese fuera al fin el camino tan ansiado por el que avanzar. Y merecemos saber todos si de verdad es o no es ese
Aunque desde aquí recuerdo una cosa. Nadie, ninguno de los expertos que durante décadas persiguen la explicación de la enfermedad que ahora sufro, ha encontrada nada relevante. Nada relevante es eso, nada relevante. Se van sabiendo detalles de aquí y allá, pero absolutamente nada que de verdad abra siquiera un camino por el que avanzar en la investigación y comprensión de la enfermedad. Eso ha sido muy frustrante tanto para los profesionales como para los millones de muertos por la enfermedad y los que la sufrimos ahora. Simplemente por esa falta de resultados histórica, el hecho de que los hongos (donde nunca jamás nadie miró) puedan ser el camino por el que seguir merece que gran parte de los expertos pongan todos sus ojos en esa dirección hasta ver si es la correcta.
A todos nos gustaría que ese fuera al fin el camino tan ansiado por el que avanzar. Y merecemos saber todos si de verdad es o no es ese. Para andar hay que empezar dando el primer paso, y en la lucha científica contra la ELA no tenemos ni eso.
Para terminar, me gustaría ampliar información sobre Luis Carrasco, porque su biografía lo merece. Catedrático de Microbiología de la UAM, se quedó ciego por una retinopatía hace dos décadas y fue capaz de ser el primer enfermo en hallar la causa de su enfermedad, hasta entonces desconocida y que afecta a 12.000 personas en España al año. Su éxito llegó en 2005, y repasando la hemeroteca encontré estas palabras suyas nada más publicar sus conclusiones tras nueve años investigándose a sí mismo: “Prefiero estar ciego y saber la causa que seguir viendo mientras la ignoro”. Ese es, en resumen, el espíritu del saber y la ciencia, ¿no?
Por cierto, la causa de su enfermedad resultaron ser los hongos, de ahí que se especializara en buscarlos de manera diseminada por el organismo y especialmente en el sistema nervioso, y que siga centrándose en ellos, ahora para intentar explicar el Alzheimer, la esclerosis múltiple o la esclerosis lateral amiotrófica. Y con la ELA puede que, con este estudio recién publicado, haya vuelto a acertar. Les toca a sus colegas seguir el trabajo y ver si hay camino por recorrer en esa dirección. Él y su equipo, seguros de estar en lo correcto, prometen seguir esos pasos.
Si así resultara de forma definitiva, Carrasco lo habría conseguido mirando hacia donde nadie había mirado nunca. Eso se llama talento, no depende de grandes presupuestos y en España lo tenemos y lo formamos de sobra. Aunque luego se le desprecie y no se le dé las oportunidades que merece. Algo que nos convierte en un país más triste y pobre en todos los sentidos. Quizá si eso no pasara, un estudio como este sería mucho más completo, tendría mayor recorrido y no sería mirado con tanto escepticismo en la escena internacional. Pero eso ya es otra historia, que me lío y empiezo a hablarles de temas que no tocaban hoy. Hoy nos tocaba mirar al futuro, no a los erróneos caminos ya recorridos.
•
•
 Desarrollo neuronal y contaminación
Desarrollo neuronal y contaminación BACTERIAS DE LAS ENCÍAS, ¿IMPLICADAS EN EL ALZHEIMER?
BACTERIAS DE LAS ENCÍAS, ¿IMPLICADAS EN EL ALZHEIMER?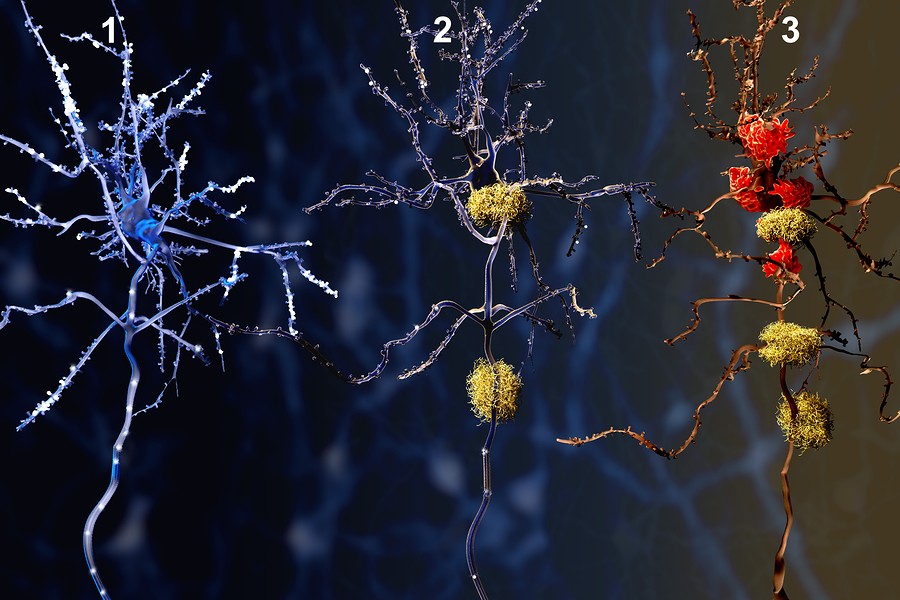
 FRENAR LA PÉRDIDA DE SINAPSIS EN EL ALZHEIMER
FRENAR LA PÉRDIDA DE SINAPSIS EN EL ALZHEIMER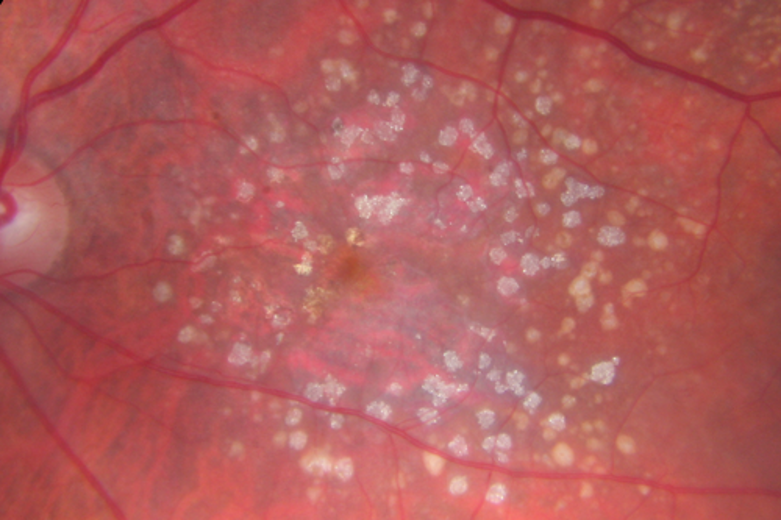
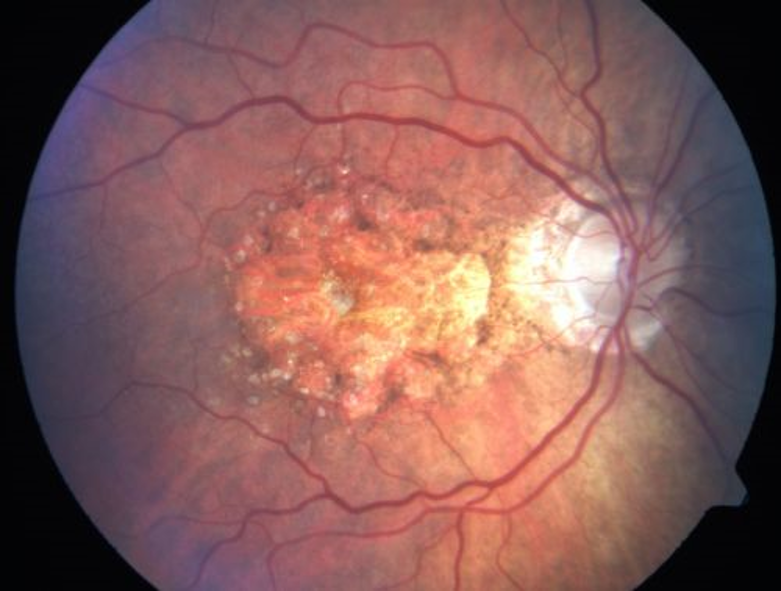 BORRAR LAS DRUSAS MACULARES PARA PREVENIR LA DMAE
BORRAR LAS DRUSAS MACULARES PARA PREVENIR LA DMAE  Alzheimer y Robert Moir
Alzheimer y Robert Moir 
 LA PROTEÍNA TAU, , TAMBIÉN SE EXPRESA EN LOS GLIOMAS
LA PROTEÍNA TAU, , TAMBIÉN SE EXPRESA EN LOS GLIOMAS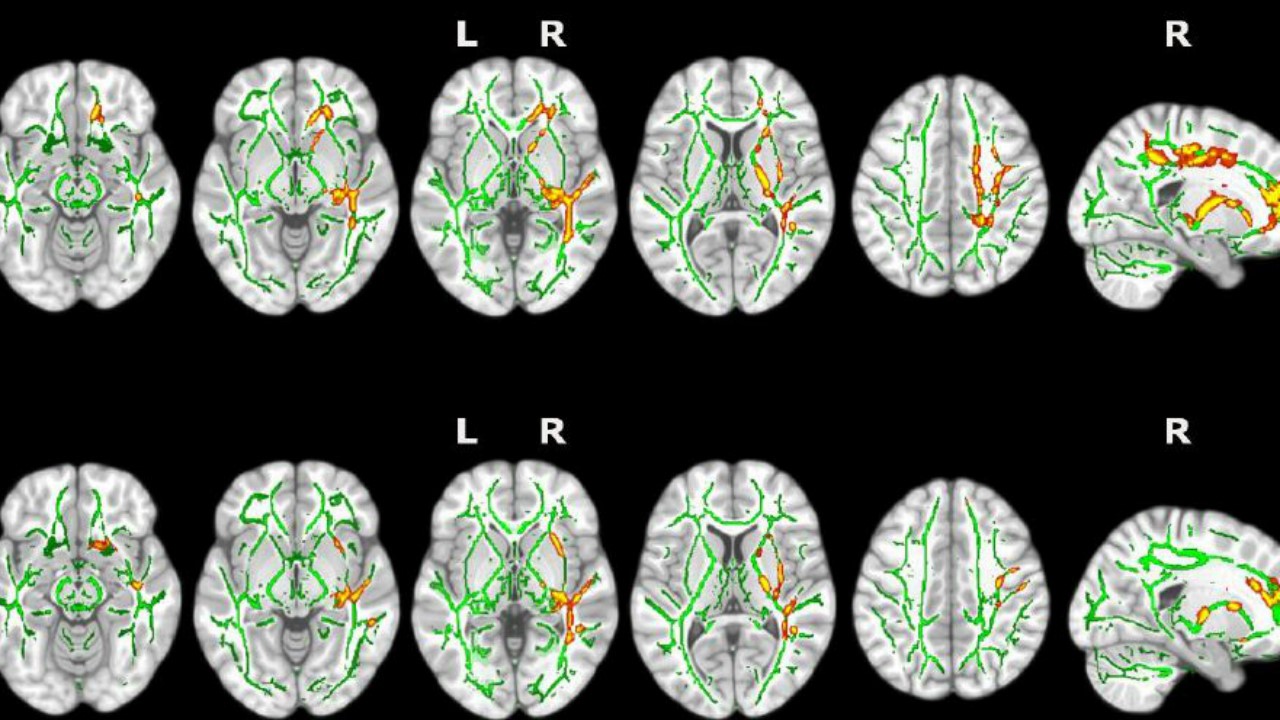 EL INSOMNIO CAUSA ALTERACIONES CEREBRALES PRECURSORAS DE ALZHEIMER
EL INSOMNIO CAUSA ALTERACIONES CEREBRALES PRECURSORAS DE ALZHEIMER