ACTUALIZACION DE LOS LINFOMAS
Se puede imaginar el lector, que este articulo es copiado de Internet, pero me parece sensato y me atrevo a copiarlo
El linfoma es un cáncer que afecta a los linfocitos que son células que forman parte del sistema inmune y ayudan a luchar frente a las infecciones
Los linfocitos se encuentran en los ganglios linfáticos y en órganos linfoides tales como la médula ósea o el bazo en los linfomas los linfocitos se reproducen de forma incontrolada dado que el tejido linfático se encuentra en todo el organismo los linfomas pueden originarse en cualquier parte del cuerpo y de ahí diseminarse a otros órganos y tejidos.
Su sintomatología es muy variada y van a depender de cada tipo. En general en la mayoría de los casos el síntoma más común es la aparición de una adenopatía que no suele ser dolorosa y que se localiza en el cuello en las axilas o las ingles. Algunos pacientes pueden presentar fiebre inexplicada, sudoración nocturna, pérdida, de peso, cansancio, picor de piel y aparición de manchas cutáneas de coloración rojiza.
Se deben diagnosticar obligatoriamente mediante una biopsia y debe ser una biopsia o una extracción de un ganglio anormal para laborar estudios de biología molecular que permitan la correcta tipificación de la enfermedad .
No hay factores de riesgo conocidos y definidos como predisponentes para el linfoma. Se les relaciona con enfermedades infecciosas por el ‘Helicobacter pylori’ o infecciones por virus de Esteinbar o con personas que estén en contacto conplagicidas, o pacientes que han sido sometidos tratamientos con inmunosupresores por trasplante de órganos sólidos pero realmente no conocemos los factores que puedan predisponer con seguridad al linfoma.
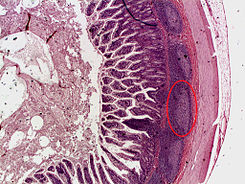
Tejido linfoide placas de Peyer en el intestino
El tejido linfoide asociado a las mucosas (MALT, por sus siglas en inglés Mucosa-associated lymphoid tissue) o folículos linfáticos es un tipo de agrupación de células linfoides sin organización o estructura, que se encuentra asociado a la mucosa y que forma parte de una serie de localizaciones linfoides repartidas por el organismo. Son un depósito de tejido linfático, incluido en fibras elásticas y músculo liso y que, a diferencia de los ganglios linfáticos, no tienen una cápsula de tejido conectivo.Los MALT se disponen de varias formas en el organismo aislados en la lámina propia y submucosa de los órganos de los sistemas digestivo, respiratorio y genitourinario, formando estructuras más complejas, asociadas con el tubo digestivo (como las amígdalas, las placas de Peyer en el intestino y el apéndice cecal) y constituyendo los órganos linfáticos como el bazo y los ganglios linfáticos. Los ganglios linfáticos estructuras encapsuladas reniformes es decir de forma parecida a un riñón, son los únicos órganos linfáticos interpuestos en el trayecto de los vasos linfáticos mayores, por lo que poseen vasos linfáticos aferentes y eferentes. Las amígdalas, el bazo y el timo tienen vasos eferentes que salen de ellos, pero que no se relacionan con vasos linfáticos aferentes.Hay cuatro tipos según el tipo de mucosaTejido linfoide asociado a los bronquios o BALT (bronchus-associated lymphoid tissue). se encuentra en la mucosa que recubre las vías respiratorias. Contiene linfocitos B y T.Tejido linfoide asociado al tubo digestivo o GALT (gut-associated lymphoid tissue). Se compone de folículos linfoides a todo lo largo del tubo gastrointestinal casi todos están aislados entre sí. Destacan las placas de Peyer, situadas en la lámina propia de la mucosa del intestino delgado, con mayor proporción en el íleon.Tejido linfoide asociado a la nariz o NALT (nose-associated lymphoid tissue).Tejido linfoide asociado a la conjuntiva o CALT (conjunctiva-associated lymphoide tissue).Existe un tipo de linfoma que se origina en los MALT y se denomina linfoma tipo MALT o linfoma de tejido linfoide asociado a la mucosa.ReferenciasOrphanet Linfoma MALT. Consultado el de septiembre de El linfoma se caracteriza por la proliferación maligna de linfocitos que constituyen las células defensivas del sistema inmunitario. El tejido linfoide se encuentra fundamentalmente en los ganglios linfáticos, por lo que los linfomas se caracterizan, generalmente, por la presencia de ganglios linfáticos de tamaño aumentado. Sin embargo, también hay células linfoides en otros muchos órganos, por lo que los linfomas pueden afectar al tubo digestivo, al bazo, al hígado, al pulmón, a la médula ósea, etc.La incidencia de esta patología es elevada, ya que cada año se diagnostican en España . nuevos casos en adultos mayores de años.
SÍNTOMAS DEL LINFOMA
En general, los linfomas se presentan como ganglios linfáticos aumentados de tamaño, que cuando aparecen en zonas accesibles como cuello, axilas o ingle se pueden palpar evidenciando su tamaño. Sin embargo, no todo aumento de ganglio tiene su origen en un linfoma. Existen diversas infecciones y otras enfermedades que provocan un aumento de ganglios linfáticos. En ocasiones, los ganglios afectados están muy internos (abdomen, mediastino) y pueden pasar desapercibidos, por lo que el diagnóstico es más difícil y sólo se consigue cuando aparecen otros síntomas (fiebre, sudoración nocturna, cansancio, pérdida importante de peso, prurito…), que obligan a realizar estudios más exhaustivos. 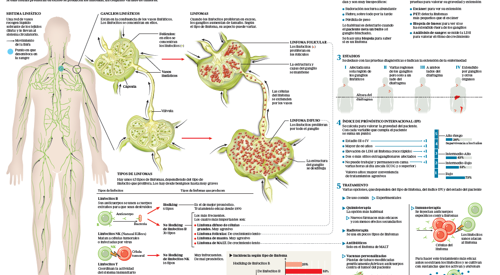
Actualmente se desconoce la causa que origina los linfomas. Suponen el % de todas las enfermedades neoplásicas hematológicas. Sin embargo, se ha constatado que el linfoma de Hodgkin es más común entre personas de a y de a años de edad. Uno de los motivos por los que podría aparecer este linfoma podría estar asociado con una infección pasada con el virus de Epstein-Barr (VEB).Por otro lado, los pacientes con infección por VIH presentan un mayor riesgo de contraer la enfermedad que la población general.Por su parte, los linfomas no Hodgkin, aunque pueden aparecer a cualquier edad, tienen una incidencia de menos del % en niños. La mayoría de los subtipos aumentan su frecuencia con la edad, siendo la media de aparición los años. Resulta algo más frecuente en varones y las causas son desconocidas.
LINFOMA NO HODGKIN
El linfoma no Hodgkin (NHL) es uno de los cánceres más comunes en los Estados Unidos, representando alrededor de 4% de todos los cánceres. Los cálculos más recientes de la Sociedad Americana Contra El Cáncer en cuanto al linfoma no Hodgkin indican que para el 2022:
Aproximadamente 80,470 personas (44,120 hombres y 36,350 mujeres) serán diagnosticadas con linfoma no Hodgkin. Esto incluye tanto adultos como niños.
Aproximadamente 20,250 personas (11,700 hombres y 8,550 mujeres) morirán debido a este cáncer.
En general, el riesgo promedio de un hombre de desarrollar linfoma no Hodgkin durante la vida es de alrededor de 1 en 42; para una mujer el riesgo es de alrededor 1 en 52. Sin embargo, el riesgo de cada persona también se puede afectar por varios .El linfoma no Hodgkin puede presentarse a cualquier edad. De hecho, es uno de los cánceres más comunes tanto en los niños, adolescentes como en los adultos jóvenes. Aun así, el riesgo de padecer linfoma no Hodgkin aumenta durante toda la vida, y más de la mitad de los pacientes tienen 65 años o más en el momento del diagnóstico. El envejecimiento de la población de las personas probablemente contribuya a un aumento en los casos de linfoma no Hodgkin durante los próximos años
¿Cuál es el pronóstico de los linfomas?
Resulta muy importante realizar un diagnóstico preciso, así como un buen estudio de extensión de la enfermedad, que nos permitan disponer de todos los datos necesarios para decidir el tratamiento más adecuado. Tradicionalmente, se ha dividido a los linfomas Agresivos o de alto grado de malignidad, cuando las células malignas crecen más rápidamente. Son más graves y necesitan tratamientos más fuertes, pero son potencialmente curables. Linfomas indolentes o de bajo grado de malignidad, cuando las células son de crecimiento más lento. Permiten una calidad de vida mejor durante años pero son muy difíciles de curar definitivamente. En la actualidad, se tiende a establecer un pronóstico diferenciado en cada caso teniendo en cuenta la variedad de linfoma, su extensión y las posibilidades terapéuticas en cada paciente concreto. Los linfomas son unos de los tumores cuya incidencia va en aumento, sobre todo la de los linfomas no hodkinianos, aunque se desconoce cuáles son las causas de este incremento.Sin embargo, también se encuentran entre los tumores con más opciones terapéuticas y, teniendo en cuenta su diversidad, entre los que más posibilidades de curación tienen actualmente.
¿Cómo se diagnostica el linfoma?
En la eficacia del tratamiento desempeña un papel fundamental la consideración terapéutica global de la enfermedad, es decir, no sólo el tratamiento que cada paciente va a necesitar en el momento del diagnóstico, sino también el que pueda requerir en el futuro ante eventuales recaídas.
¿Cómo se trata el linfoma?
La quimioterapia en la mayor parte de las ocasiones permite tratamientos prolongados sin necesidad de ingreso hospitalario, con la consiguiente comodidad para el paciente y la disminución del coste económico.
En ocasiones es necesario recurrir al Trasplante de Progenitores Hematopoyéticos.
Asesinas naturales, al rescate de los linfomas CD+
Los pacientes con linfoma CD+ en recaída o refractarios al tratamiento convencional podrían tener una nueva oportunidad terapéutica. 
Esta inmunoterapia celular se ha ensayado en pacientes con linfomas CD+ (Hodgkin y linfomas T) refractarios a Brentuximab Vedotin. Sonia Moreno. MadridMar, // –
La administración de células asesinas naturales (NK) junto a un anticuerpo biespecífico -AFM, diseñado para unirse a CD A en las células NK y a CD en las células tumorales- puede convertirse en un tratamiento útil en pacientes con linfoma T CD+ en recaída, una enfermedad para la que no hay muchas opciones terapéuticas. Un equipo del Centro del Cáncer M. D. Anderson de la Universidad de Texas ha presentado en la reciente reunión de la Asociación Estadounidense para la Investigación del Cáncer (AACR) resultados positivos de un estudio piloto con esta estrategia.
El investigador principal del ensayo, Yago Nieto, profesor en el Departamento de Trasplante de Células Madre y Terapia Celular del citado centro estadounidense, detalla a este medio que el tratamiento se ha ensayado en “linfomas CD+ (Hodgkin y linfomas T) refractarios a brentuximab vedotin”.  Yago Nieto, del MD Anderson de la Universidad de Texas.
Yago Nieto, del MD Anderson de la Universidad de Texas.
Según ha expuesto Nieto, los pacientes, de edades , estaban muy pretratados y contaban con un número mediano de siete líneas de tratamiento previas. “Dos tercios de ellos habían ya recibido un trasplante de células stem. Todos los pacientes se encontraban en progresión tumoral al reclutamiento”.
Respuesta global y completa
El tratamiento obtuvo un % de respuesta global en el conjunto del estudio y el % en la cohorte tratada a la dosis recomendada para fase II; la tasa de respuestas completas fue del % en el global del estudio y del % en la cohorte que recibió la dosis recomendada. Dos pacientes se mantienen en respuesta completa sin tratamiento posterior meses después. El especialista destaca la excelente tolerancia del tratamiento. “No vimos ningún caso de síndrome de liberación de citocinas, neurotoxicidad o enfermedad injerto contra hospedador (EICH). La incidencia de reacciones infusionales fue baja (%). La mielotoxicidad que vimos (neutropenia y trombopenia) era la esperable con la combinación de fludarabina y ciclofosfamida necesaria para supresión inmunitaria (linfodepleción) del paciente, pero no hallamos ningún caso de fiebre neutropénica ni hemorragia. Hemos tratado a pacientes con dos ciclos de AFM-NK. Hasta ahora hemos intentado consolidar las respuestas completas con un trasplante cuando era posible. Ahora vamos a empezar a usar cuatro ciclos para explorar el potencial curativo del tratamiento por sí solo”, anuncia. Un anticuerpo afín a dos antígenos El anticuerpo AFM tiene especificidad por CD en las células tumorales y por CDA en las células NK. Es tetravalente, lo que le confiere alta afinidad por ambos antígenos. Las NK se obtuvieron de sangre de cordón puesto que aquí están más presentes (%) que en sangre periférica (%). “La sangre de cordón es rápidamente accesible, a diferencia de la sangre periférica de donante. Tras elegir una unidad de sangre de cordón de nuestro banco (sin considerar la compatibilidad HLA con el paciente, que en el caso de las NK no importa), las fabricamos en nuestro laboratorio GMP, empezando por la separación inmuno magnética de las NK, seguido de la preactivación de las NK en presencia de citocinas (IL-, IL- e IL-), su expansión en presencia de células feeder (que resulta en un número final de NK más de . veces mayor al inicial) y finalmente, de la incubación con AFM durante una hora. Todo el proceso lleva días. Es decir, en solo dos semanas pasamos de elegir un cordón a infundir AFM-NK al paciente”, describe. “Además, las NK de cordón son más estimulables para su proliferación y persisten más tiempo en el paciente que las de sangre periférica. En cambio, presentan un fenotipo menos activado (menos citotóxico), pero esto se compensa mediante la preactivación con citocinas que induce en ellas un estado activado de memoria”, continúa. La gran ventaja de las células NK frente a las células T es su uso alogénico “sin riesgo de EICH, ni necesidad de intervenciones genéticas laboriosas para neutralizar los receptores que median en esta enfermedad (como es necesario hacer con las CAR-T alogénicas). La incubación con AFM confiere a las NK una especificidad y una activación muy similar a la que experimentan cuando se les implanta un receptor quimérico artificial (CAR), pero el proceso es bastante más rápido y a mucho menor coste. Por otra parte, el perfil de efectos secundarios de las células NK es claramente mejor que el de las CAR-T”. SGraduada en Enfermería, Universidad de Zaragoza.Graduada en Enfermería, Universidad de Zaragoza.Graduada en Enfermería, Universidad de Zaragoza.Graduada en Enfermería, Universidad de Zaragoza.Graduada en Enfermería, Universidad de Valencia.Graduada en Enfermería, Universidad de Zaragoza.ResumenEl Linfoma de Hodgkin es un tipo de cáncer que afecta al sistema linfático. La etiología es desconocida. El principal síntoma es la inflamación de un ganglio sin causa conocida. Asimismo, son frecuentes los denominados “Síntomas B” pérdida de peso superior al % en los últimos seis meses sin causa determinada, fiebre vespertina y sudoración nocturna. El diagnóstico debe realizarse a través de una biopsia de un ganglio sospechoso.El tratamiento dependerá del tipo de linfoma de Hodgkin y de su diseminación, siendo frecuente la administración conjunta de quimioterapia y radioterapia. Finalmente, el linfoma de Hodgkin es el cáncer con mayor tasa de curación existente, siendo esta superior al %.
Palabras clave Linfoma de Hodgkin, sintomatología, tratamiento, células Reed-Sternberg.AbstractEnviar artículo para publicarHodgkin lymphoma is a type of cancer that affects the lymphatic system. The etiology is unknown. The main symptom is a swollen node with no known cause. Likewise, the so-called «B symptoms» are frequent weight loss greater than % in the last six months without a specific cause, evening fever and night sweats. The diagnosis must be made through a biopsy of a suspicious node. Treatment will depend on the type of LH and its spread, with the joint administration of chemotherapy and radiotherapy being frequent. Finally, Hodgkin’s Lymphoma is the cancer with the highest existing cure rate, being over %.Keywords Hodgkin Lymphoma, symptomatology, treatment, Reed-Sternberg cells.IntroducciónEl linfoma de Hodgkin es un tipo de cáncer que afecta al sistema linfático.El sistema linfático está formado por un conjunto de órganos, ganglios linfáticos, conductos y vasos sanguíneos encargados de producir y transportar la linfa desde los tejidos hasta la sangre.Este sistema está compuesto por células llamadas linfocitos (glóbulos blancos). Y encontramos tiposLinfocitos B (células B) encargados de producir anticuerpos.Linfocitos T (células T) encargados de producir la respuesta inmunitaria ,.El linfoma de Hodgkin se produce cuando los mecanismos de control de los linfocitos se alteran, iniciándose así una división celular alterada e incontrolada .La esperanza de vida del Linfoma de Hodgkin dependerá del tipo de linfoma de Hodgkin, de su diseminación y de la respuesta ante el tratamiento. Sin embargo, es una de las enfermedades que presentan mayor tasa de curación, por encima del %.Causas y factores de riesgoLa causa de esta patología es desconocida. Hay datos que indican una posible relación de linfoma de Hodgkin con el virus Epstein-Barr (VEB), pero hasta el momento no se ha establecido una prueba definitiva. Tampoco se ha demostrado una asociación con factores laborales o ambientales, ni relación con exposición a radiación, manejo de productos químicos ni biocidas ,.Hay datos epidemiológicos que evidencian la posibilidad de que exista cierta predisposición genética para desarrollar linfoma de Hodgkin. Familiares de primer grado que padecen esta enfermedad presentan un riesgo de hasta cinco veces mayor; los gemelos monocigóticos de una persona con linfoma presentan una probabilidad casi veces mayor de padecer la enfermedad respecto a los gemelos dicigóticos. Sin embargo, esto no significa que el linfoma de Hodgkin sea una enfermedad hereditaria .La incidencia de Linfoma de Hodgkin en Europa es de , a casos por cada habitantes ; extrapolando estos datos cada año se producen en España casos nuevos . El linfoma de Hodgkin puede afectar a personas de cualquier edad, siendo más frecuente entre los – años y en mayores de años .Signos y síntomasPor lo general, la primera señal de Linfoma de Hodgkin es la inflamación de un ganglio linfático sin causa conocida. Preferentemente, se localizan en región cervical, seguido de región axilar e inguinal. Esta enfermedad puede diseminarse a ganglios linfáticos cercanos e incluso a órganos como el bazo, hígado y médula ósea, entre otros ,.Cuando el crecimiento ganglionar se encuentra a nivel abdominal o torácico se producen una serie de síntomas por compresión de estos ganglios sobre estructuras anatómicas. Así, encontramos tos y dificultad respiratoria (en caso de comprimir tráquea o bronquios), dolor abdominal o de espalda (si afectación de ganglios abdominales) .Alrededor del % de los casos, los pacientes presentan los llamados “síntomas B” pérdida de peso superior al % en los últimos seis meses sin causa determinada, fiebre vespertina y sudoración nocturna .Asimismo, los pacientes pueden presentar picores cutáneos generalizados y lesiones de rascado de varios meses de evolución (-% de los casos).Un síntoma clásico, pero poco frecuente, es la aparición de dolor en los ganglios linfáticos tras la ingesta de alcohol, denominado signo de Oster .
Diagnóstico
El diagnóstico del Linfoma de Hodgkin debe establecerse tras una biopsia de ganglio linfático sospechoso .La punción aspiración de un ganglio linfático inflamado, no es una técnica válida para el diagnóstico de los linfomas ya que no permite visualizar la estructura del ganglio al microscopio .Cuando no existen ganglios externos inflamados puede realizarse el diagnóstico a través de biopsia mediante punción con aguja gruesa .Si tras los exámenes se diagnostica Linfoma de Hodgkin, se llevarán a cabo otros exámenes para ver hasta dónde se ha diseminado el cáncer, es decir, estadificación. Esto ayuda a seleccionar el tratamiento correcto y a guiar el seguimiento. En este sentido, son necesarios una analítica de sangre (niveles de proteínas, pruebas de función hepática y renal, nivel de ácido úrico), biopsia de médula ósea, PET/TAC y conteo sanguíneo completo (para ver si hay anemia y conteo de leucocitos) ,.
Estadios del linfoma de Hodgkin
Estadio I el linfoma afecta a una sola región ganglionar o afecta a un solo órgano fuera del sistema linfático.
Estadio II existen dos o más regiones ganglionares afectas en el mismo lado del diafragma.
Estadio III se afectan varias regiones ganglionares a ambos lados del diafragma.Estadio IV se afectan uno o más territorios extra ganglionares (médula ósea, hígado, pulmones) ,.El estadio de la clasificación de Ann Arbor se acompaña de las letras A, B, E o SA no existen síntomas B en el momento del diagnóstico. B existen síntomas B.E existe afectación de una región extra ganglionar.S existe afectación a nivel del bazo .Tipos de linfoma de Hodgkin Según la Organización Mundial de la Salud (OMS) el linfoma de Hodgkin lo podemos dividir principalmente en dos tipos Linfoma de Hodgkin clásico (% casos) y Linfoma de Hodgkin con predominio linfocitario nodular (%) ,.Las células cancerosas del Linfoma de Hodgkin clásico (cHL) se denominan células Reed-Sternberg. Los ganglios linfáticos agrandados en personas con cHL tienen un pequeño número de células Reed-Sternberg con células inmunitarias normales circundantes.
Subtipos del linfoma de Hodgkin
El Linfoma de Hodgkin clásico tiene cuatro subtipos Linfoma de Hodgkin con esclerosis nodular subtipo más común. Tiende a originarse en los ganglios linfáticos del cuello o tórax. Linfoma de Hodgkin con celularidad mixta se puede originar en cualquier ganglio y principalmente en la mitad superior del cuerpo. Linfoma de Hodgkin con predominio linfocitario no suele presentarse en más de un ganglio linfático. Linfoma de Hodgkin con depleción linfocitaria es más agresivo que otros subtipos y es probable que se detecte en estadios avanzados de la enfermedad. Suele afectar a ganglios linfáticos de abdomen, bazo, hígado y médula ósea . Las células cancerosas del Linfoma de Hodgkin con predominio linfocitario nodular (NLPHL) son grandes y se denominan células popcorn o palomitas de maíz, debido a su aspecto. Estas células son variantes de las células Reed-Sternberg. Por lo general, este tipo de linfoma de Hodgkin se origina en los ganglios linfáticos del cuello y brazo. Suele crecer más rápidamente que el cHL y presenta diferente tratamiento .
Tratamiento
El tratamiento del linfoma de Hodgkin depende del tipo de linfoma de Hodgkin, del estadío en que se encuentre, de la edad y otras cuestiones médicas, así como de otros factores (pérdida de peso, sudoración nocturna, fiebre).
Tratamiento de primera línea
El esquema quimioterápico estándar es el ABVD (combinación de adriamicina, bleomicina, vinblastina y dacarbacina). Se administra vía intravenosa en un ciclo que se repite cada días con dos administraciones, una en el día y otra en el día .Pacientes diagnosticados en estadíos iniciales
Sin factores de mal pronóstico el tratamiento recomendado es la administración de dos ciclos de ABVD y radioterapia complementaria sobre campos afectos (Gys).
Con factores de mal pronóstico el tratamiento recomendado es la administración de cuatro ciclos de ABVD y radioterapia complementaria (Gys).
Pacientes diagnosticados en estadíos avanzados el tratamiento se basa en seis ciclos de ABVD (adriamicina, bleomicina, vinblastina y dacarbacina). Si el PET/TAC al finalizar la quimioterapia es negativo, no es necesario administrar radioterapia complementaria .
Tratamiento de segunda línea y posteriores
Entre un -% de los pacientes no responden al tratamiento de primera línea y un % de los pacientes, después de conseguir una remisión completa, recaen de la enfermedad. En estas situaciones, los pacientes son tratados con esquemas de quimioterapia de segunda línea que son más intensivos que los de la primera. Si el paciente responde bien a este tratamiento se consolida con quimioterapia a dosis altas y trasplante autólogo de progenitores hematopoyéticos.
En la actualidad, se dispone de nuevos fármacos indicados en pacientes que están en recaída o son refractarios al tratamiento previo. Entre ellos tenemos
Brentuximab vedotina efectivo en pacientes en recaída tras un trasplante autólogo o en pacientes que han fracasado a dos líneas diferentes de quimioterapia previa y no son candidatos a un trasplante autólogo.
Inhibidores de Checkpoint (nivolumab, pembrolizumab) en aquellos que recaen tras un trasplante autólogo y fracasan al tratamiento con brentuximab vedotina .
El linfoma de Hodgkin es un tipo de cáncer del sistema linfático. Su sintomatología es silente, por lo que la mayor parte de los diagnósticos proceden de consultas por inflamación de un ganglio. El tratamiento depende de distintos factores, siendo el Linfoma de Hodgkin el tipo de cáncer con mayor tasa de curación.
Bibliografía
Sara Pérez Morata Ocronos. Vol. III. Nº – Diciembre . Pág. Inicial Vol. III;nº Mayo Clinic [Internet]. . [Citado el noviembre ]. Disponible en https//www.mayoclinic. org/es-es/diseases- conditions/hodgkins- lymphoma/symptoms- causes/syc-
¿Qué es el Linfoma de Hodgkin? Cancer.org [Internet]. [Actualizado mayo ; citado noviembre ]. Disponible en https//www.cancer.org/ es/cancer/linfoma-hodgkin /acerca/que-es-enfermedad -de-hodgkin.html AECC Asociación Española Contra el Cáncer [Internet]. Madrid AECC [actualizado ; citado noviembre ]. Disponible en https//www.aecc. es/es/todo-sobre-cancer/ tipos-cancer/linfoma-tipo-hodgkin? gclid=EAIaIQob ChMIW YsPQA IVAiICRQkgIq EAAYAiAAEgJ JfD_BwE Fundación Josep Carreras. Linfoma de Hodgkin [Internet]. Barcelona [Actualizado noviembre , citado noviembre ]. Disponible en https//www.fcarreras. org/es/linfomadehodgkin
A.Rueda. Linfoma de Hodgkin. SEOM Sociedad Española de Oncología Médica [Internet]. Madrid SEOM [Actualizado enero ; Citado noviembre ]. Disponible enhttps//seom.org/info- sobre-el-cancer/linfoma -hodgkin?showall=
Medline Plus [Internet]. Bethesda (MD) U.S. National Library of Medicine [actualizado noviembre ; citado noviembre ]. Disponible en https//medlineplus .gov/spanish/ ency/article/.htm







 El perfil farmacocinético de ▼ Refixia® favorece la individualización del tratamiento de la hemofilia B proporcionando protección frente al sangrado con un reducido número de infusiones intravenosas¹
El perfil farmacocinético de ▼ Refixia® favorece la individualización del tratamiento de la hemofilia B proporcionando protección frente al sangrado con un reducido número de infusiones intravenosas¹ Kyowa Kirin pone en marcha un proyecto para mejorar el abordaje de los pacientes con Micosis Fungoide y Síndrome de Sézary
Kyowa Kirin pone en marcha un proyecto para mejorar el abordaje de los pacientes con Micosis Fungoide y Síndrome de Sézary La contribución desde la farmacia en la lucha contra el cambio climático
La contribución desde la farmacia en la lucha contra el cambio climático La epilepsia farmacorresistente: cuando los tratamientos convencionales no son suficientes
La epilepsia farmacorresistente: cuando los tratamientos convencionales no son suficientes
. Foto: LUIS CAMACHO")