FISIOPATOLOGÍA DE LOS TRANSPORTADORES DE GLUTAMATO Y DE GLICINA:
Los mecanismos de regulación del glutamato y glicina para que hagan sus efectos neurotransmisores es tan compleja, que cuesta trabajo entender.
La homeostasis de la transición de señales en sí ya es un problema de entendimiento, enzarzar está propiedades de algunos neurotransmisores dentro de la complejidad de un cerebro que además piensa a mí personalmente me cuesta un trabajo extraordinario.
La neurofisiología de estos componentes son entendibles, pero cuando actúan en un medio inesperado que casi siempre termina en inflamación, ya es inimaginable
Los aminoácidos glutamato y glicina, aparte de su papel en la síntesis de proteínas, son dos neurotransmisores fundamentales en el sistema nervioso central de los mamíferos. El primero es ubicuo y está implicado en vías excitatorias de la neocorteza, la retina y el cerebelo, y el segundo está asociado a vías inhibitorias de zonas caudales del cerebro. Sin embargo, ambos comparten su manera de actuar al integrarse en el funcionamiento de los receptores de glutamato del tipo NMDA, fundamentales en la regulación de sistemas motores, sensitivos y cognitivos.
El conocimiento de la forma molecular de actuar de los transportadores de glutamato y de glicina está permitiendo la identificación y el desarrollo de nuevas estrategias terapéuticas para patologías como las descritas y el desarrollo de nuevos fármacos.)
El glutamato, además de sustrato en la síntesis de proteínas, es el neurotransmisor excitador más abundante en el cerebro. La neurotransmisión glutamatérgica está implicada y regula sistemas motores, sensitivos y cognitivos. Igualmente, desempeña un papel primordial en la plasticidad sináptica. La concentración de glutamato en el cerebro es entre 1.000 a 10.000 veces superior a la de las aminas biógenas [1,2]. Sin embargo, unos niveles anormalmente elevados de glutamato provocan la muerte neuronal por neurotoxicidad [3–5].
Para que la neurotransmisión mediada por el glutamato sea eficiente, sus niveles en el interior de las neuronas y de las células de la glía, y principalmente en el espacio intercelular, deben estar muy finamente regulados y mantenidos en concentraciones adecuadas.
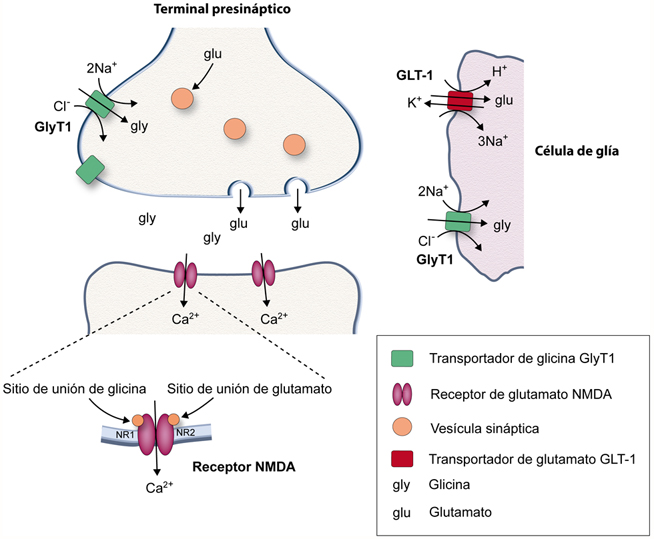
Este proceso se lleva a cabo mediante una serie de transportadores localizados en la membrana plasmática de las neuronas y las células gliales [2,6–8] (Fig. 1).
Concretamente, existen cinco transportadores de glutamato (EAAT) que difieren en su secuencia aminoacídica, capacidad de transporte para glutamato, afinidad, velocidad de transporte, conductancia al cloruro y, sobre todo, localización celular. Todos comparten la dependencia del sodio en el ciclo de transporte, aparte de otros iones adicionales. Los genes que codifican los transportadores en los humanos son SLC1A3, SLC1A2, SLC1A1, SLC1A6 y SLC1A7, correspondientes a las proteínas EAAT1, EAAT2, EAAT3, EAAT4 y EAAT5 (las tres primeras llamadas GLAST, GLT-1 y EAAC1 en los roedores). Además, los transportadores EAAT1, 2 y 3 presentan variantes de splicing alternativo en el procesamiento de sus ARN mensajeros que difieren en sus extremos amino o carboxilo. Esta característica confiere a cada variante posibilidades diferentes para interaccionar con otras proteínas, aunque no hay evidencias de diferencias funcionales entre ellas [9,10].
La concentración estimada de glutamato en el cerebro está en el rango de 5-15 mM [2]. Sin embargo, este neurotransmisor no está distribuido uniformemente. La concentración extracelular de glutamato se mantiene probablemente por debajo de 100 nM (entre 10 y 100 nM) [11,12], a diferencia de concentraciones intracelulares de alrededor de 10 mM en el citoplasma [13] y de 60 mM en el interior de las vesículas sinápticas [14].
Los transportadores de glutamato, funcionando en situaciones fisiológicas, son capaces de mantener gradientes de concentración a través de la membrana plasmática hasta de un millón de veces. El mantenimiento de estos gradientes en condiciones celulares de reposo requiere un esfuerzo termodinámico de enormes proporciones llevado a cabo por el funcionamiento de los transportadores de glutamato que operan a través de ciclos de transporte con una estequiometria de una molécula de glutamato, tres de iones sodio y un protón, que se contraponen al movimiento de un ion potasio [15]
(Fig. 1). El funcionamiento de todos los transportadores de glutamato lleva aparejado un flujo termodinámico de iones cloruro no acoplado a través del transportador [13,15,16]. En este sentido, en los transportadores neuronales EAAT4 y EAAT5, este flujo de iones cloruro es muy acusado, y ambos funcionan como verdaderos canales de cloruro. El resto de los transportadores EAAT1-3 presentan un flujo desacoplado de cloruro mucho más limitado [17–19]. En muchos casos, el significado fisiológico de estas corrientes desacopladas de cloruro no está aún bien establecido.
Figura 1. Esquema de una sinapsis glutamatérgica excitatoria. Se representan los receptores postsinápticos del tipo NMDA; los transportadores de glicina del tipo GlyT1 y el transportador de glutamato del tipo GLT-1. El glutamato almacenado en vesículas sinápticas del terminal presináptico se libera al espacio intersináptico y se une, junto con la glicina, a receptores NMDA, provocando la entrada de calcio al terminal postsináptico.
La localización regional y celular de los trasportadores de glutamato es muy específica, lo que hace que su funcionamiento en condiciones fisiológicas no resulte redundante, como podría parecer en un principio. EAAT1 (GLAST) y EAAT2 (GLT-1) se expresan de forma abundante y ubicua en el cerebro, aunque GLT-1 se encuentra principalmente en la neocorteza y el hipocampo, y GLAST en el cerebelo [2]. EAAT3, aunque ubicuo en el sistema nervioso central, se localiza principalmente en el soma y las dendritas de las neuronas del hipocampo, así como en el cerebelo y los ganglios basales. EAAT4 tiene una localización preferente en las dendritas de las células de Purkinje del cerebelo [20–23], y EAAT5 es una proteína confinada a terminales presinápticos de las células bipolares de la retina y fotorreceptores, con muy poca presencia en el resto del sistema nervioso [24,25].
GLT-1, que probablemente represente el 1% de todas las proteínas del cerebro, se expresa mayoritariamente en los astrocitos y es responsable de la mayor cantidad del transporte de glutamato al interior celular [26–28]. Por ello, la deleción genética de GLT-1 tiene como resultado la aparición de convulsiones en las primeras etapas del desarrollo y la muerte prematura de los animales [29]. Aunque en mucha menor proporción que en los astrocitos, se ha demostrado expresión presináptica de GLT-1 [30]. Este transportador localizado presinápticamente podría actuar reponiendo las reservas presinápticas de glutamato como vía alternativa al ciclo glutamato-glutamina. Tradicionalmente se ha postulado que el glutamato almacenado en las neuronas se recupera del entorno mediante el transportador GLT-1 localizado en los astrocitos; dentro de éstos es convertido en glutamina, la cual se transporta a las neuronas presinápticas, y de nuevo es transformado en glutamato, el cual se almacena en las vesículas sinápticas a la espera de ser liberado como neurotransmisor. GLAST se expresa en los astrocitos y oligodendrocitos del sistema nervioso y, a diferencia de lo que ocurre con GLT-1, los animales con la deleción genética de esta proteína muestran pocas anormalidades funcionales [31].
La mayoría de los transportadores de glutamato (EAAT1 y EAAT2) forman complejos homotriméricos de tres subunidades idénticas, si bien algunos son heterotrímeros (EAAT3 y EAAT4) [32,33]. Parece que cada subunidad puede funcionar de forma independiente en cuanto al transporte, pero siempre en forma de trímeros [34]. A lo largo del ciclo de transporte, cada subunidad sufre varios cambios conformacionales. En primer lugar, en el espacio extracelular se une una molécula de glutamato, así como tres iones sodio y un protón. En este momento, el transportador sufre un cambio conformacional adoptando una forma de ‘cara hacia adentro’ que permite liberar estas sustancias en el citoplasma. A continuación, se une un ion potasio desde el interior, el cual es liberado al espacio extracelular mediante otro cambio conformacional de la proteína, volviendo al estado de ‘cara hacia afuera’ [35,36]. Cada protómero del complejo tiene una estructura peculiar formada por ocho regiones transmembrana orientadas no perpendicularmente a ella [7,37]. Mediante diferentes técnicas, principalmente mutagénesis dirigida, en los últimos años se ha podido determinar el sitio concreto de la proteína donde se unen tanto el sustrato como cada uno de los iones acoplantes (Na+1, 2 y 3, K+, H+), así como inhibidores y bloqueantes del transportador [38].
La actividad de los transportadores de glutamato puede ser alterada por agentes farmacológicos, mediadores endógenos y bastantes sistemas reguladores celulares. El primer grupo reúne a sustancias que pueden actuar como inhibidores transportables por el transportador (inhibidores competitivos) o como bloqueantes del transportador no transportables [8]. Los inhibidores transportables suelen ser análogos estructurales del glutamato o del aspartato, los cuales son transportados por la proteína, compitiendo así por el transporte de glutamato. Además del aspartato, en este grupo se encuentran el metilglutamato y una serie de derivados del anillo de la pirrolidina en forma de ácidos dicarboxílicos. Dentro del grupo de los bloqueantes no transportables del transportador destacan el kainato y su derivado, el dihidrokainato. Ambos compuestos reaccionan específicamente con el transportador GLT-1, aunque también se unen a receptores de glutamato del tipo kainato. Los compuestos denominados TBOA (DL-treo-beta-benzoilaspartato) se unen a los cinco tipos de transportadores de glutamato, variando en sus afinidades. Lo más interesante de estos compuestos es su nula afinidad por los receptores de glutamato. En la actualidad se están desarrollando una serie de compuestos bloqueantes específicos para los cinco tipos de transportadores de glutamato.
La neurotransmisión glutamatérgica, mantiene el equilibrio entre , cantidad de glutamato y de sus receptores.
En las sinapsis en las que hay un número importante de transportadores, el glutamato se retira rápidamente del espacio intercelular; sin embargo, en las sinapsis con una baja densidad de transportadores, el glutamato difunde hacia otras sinapsis cercanas, alterando su funcionamiento. Estos transportadores están sometidos a procesos de tráfico intracelular regulados por diversas vías de señalización. Desde hace tiempo se sabe que la proteincinasa C (PKC) tiene efectos diversos sobre los transportadores de glutamato, dependiendo del tipo celular en el que se expresen, lo que indica que la regulación del transporte de glutamato por la PKC es compleja en muchos sentidos. Y no sólo depende del tipo celular, sino también del tipo de isoforma de PKC y de la forma en que ésta se active [39,40]. Circunscribiéndonos a GLT-1, mayoritaria en los astrocitos, aunque también presente en las neuronas, procesos de fosforilación de la proteína regulan la cantidad de ésta presente en la superficie de la membrana plasmática [41]. Recientemente se ha demostrado que GLT-1 se encuentra en forma de agrupaciones dinámicas que cambian dependiendo de la actividad neuronal [42]. El número de transportadores en la membrana en un momento determinado refleja el equilibrio entre su inserción y su retirada. La endocitosis de GLT-1 depende de la ubiquitinación del transportador inducida por la activación de la PKC y mediada por la ubiquitina ligasa Nedd4-2 [43,44]. Recientemente, nuestro grupo ha demostrado que el reciclaje de GLT-1 a través de endosomas hacia y desde la membrana está influido por su actividad catalítica inducida por el propio glutamato. El mecanismo molecular a través del cual opera este proceso implica que el glutamato promueve el reclutamiento de la proteína adaptadora β-arrestina 1 en el proceso endocítico, la cual a su vez captura Nedd4-2 en la membrana [45]. Todos estos eventos preceden y son necesarios para la internalización del transportador [46]. Otras cinasas, como la GSK3β, que desempeña un papel importante en múltiples procesos celulares, como el desarrollo neuronal o la plasticidad sináptica, también influyen en el tráfico intracelular de los transportadores de glutamato [47].
Implicación de los transportadores de glutamato en estados patológicos
Isquemia cerebral
La isquemia es un proceso que impide el suministro de glucosa y de oxígeno al cerebro. Ambos hechos tienen una ingente cantidad de consecuencias en el metabolismo y otros muchos procesos. Lo más evidente es una disminución muy drástica en la producción de energía en forma de trifosfato de adenosina (ATP), tanto por la vía glucolítica como por la aerobia [48]. La parada de actividad de la Na+-K+-ATPasa colapsa los gradientes de Na+ y de K+ a través de la membrana plasmática de las neuronas y de las células gliales. Puesto que los transportadores de glutamato dependen para su funcionamiento de ambos iones, un proceso de infarto o isquemia cerebral reduce drásticamente el funcionamiento de los transportadores y, como consecuencia, las concentraciones de glutamato extracelular aumentan rápidamente [49,50]. Se ha calculado que el glutamato puede variar desde concentraciones en el orden de nanomoles hasta concentraciones del orden de micromoles, lo que implica un impacto importante en su interacción con receptores de glutamato. La liberación masiva de glutamato inicia una cascada de acontecimientos que terminan en la muerte neuronal a la cual contribuyen en mayor o menor medida procesos de apoptosis, necrosis, inflamación y estrés oxidativo [51].
A este proceso de aumento de concentraciones extracelulares de glutamato mediante la reversión de su transporte influyen los transportadores GLAST (EAAT1), GLT-1 (EAAT2) y EAAT3 [52]. GLT-1 es especialmente relevante, porque es mayoritario en los astrocitos, mientras que en las neuronas, aunque minoritario, la concentración de glutamato en su citoplasma es mucho más elevada que la correspondiente en las células gliales. Por otra parte, se ha demostrado que un proceso de isquemia prolongado modifica los patrones de expresión de los transportadores indicados.
Dolor crónico
Tal como se ha descrito anteriormente, TBOA es un bloqueante de los transportadores de glutamato que se manifiesta igualmente efectivo inhibiendo tanto el proceso de recaptura como el funcionamiento reverso. En condiciones fisiológicas, el tratamiento de TBOA a animales de experimentación induce respuestas nociceptivas de forma dependiente de la dosis. Estos resultados son la consecuencia de elevar las concentraciones de glutamato que llevarían a una sobreestimulación de los receptores de glutamato presentes en neuronas sensitivas de la médula espinal. De cualquier manera, los resultados con tratamiento de TBOA dependen de las condiciones. En condiciones fisiológicas, TBOA induce dolor, mientras que en modelos de dolor crónico, la administración de TBOA lo alivia. La explicación para estos resultados contradictorios es difícil y hace complicado sacar conclusiones con fines terapéuticos [8].
Enfermedades neurodegenerativas
Desde hace tiempo, evidencias experimentales de estudios en muestras de cerebro post mortem demuestran una reducción significativa de la cantidad de GLT-1 en la corteza motora y la médula espinal en pacientes con esclerosis lateral amiotrófica, lo que lleva a un aumento importante de las concentraciones de glutamato en el líquido cefalorraquídeo [53,54]. Ello puede contribuir a la desaparición por neurotoxicidad de motoneuronas de la médula espinal. La pérdida del transportador GLT-1 parece ser selectiva porque otros transportadores, como EAAT1 y EAAT3, se mantienen sin sufrir variaciones [53]. En este sentido, hay que señalar que uno de los genes implicados en la enfermedad es SOD1, que codifica la superóxido dismutasa, una enzima implicada en la inactivación de radicales libres a los que GLT-1 es especialmente susceptible [55].
Se ha demostrado igualmente una disminución relevante de GLT-1 en la corteza cerebral motora de pacientes con enfermedad de Alzheimer. Más concretamente, de las variantes génicas de GLT-1, la que parece más afectada es GLT-1a. De cualquier manera, el resultado sería un aumento en la concentración extracelular de glutamato y el inicio de un proceso de neurotoxicidad. Un tema de actualidad e interés es intentar correlacionar tipos celulares, isoformas de GLT-1 y regiones de especial vulnerabilidad en el cerebro de pacientes con enfermedad de Alzheimer [56,57].
Desde hace algunos años, el concepto de muerte celular por excitotoxicidad se aplica no sólo a la muerte neuronal, sino que se ha extendido a células de la glía, incluyendo los astrocitos, los oligodendrocitos y la microglía. Por este motivo, se propuso que la muerte de oligodendrocitos por neurotoxicidad podría estar involucrada en la patogenia de enfermedades desmielinizantes caracterizadas por la destrucción de mielina [58,59]. Se ha demostrado una disminución importante en las cantidades y actividad de los transportadores gliales de glutamato GLT-1 y GLAST en animales con encefalomielitis autoinmune experimental [60], un modelo animal que reproduce las características clínicas y patológicas de la esclerosis múltiple [61]. Ello hace pensar que en esta enfermedad se puede dar un proceso como los descritos anteriormente, con el aumento de las concentraciones extracelulares de glutamato hasta niveles excitotóxicos, que contribuya al daño celular y al proceso de astrogliosis encontrados en la esclerosis múltiple [62,63].
Transportadores de glutamato como potenciales dianas terapéuticas
Además de las implicaciones de los transportadores de glutamato en las condiciones patológicas reseñadas, a lo largo de los últimos años se los ha involucrado en las enfermedades de Huntington y de Parkinson, la epilepsia, la esquizofrenia, los trastornos obsesivos compulsivos, la patogenia en los gliomas, la ataxia episódica y algunas enfermedades metabólicas, como la aminoaciduria dicarboxílica. Debido a la complejidad que presentan los transportadores de glutamato tanto en su distribución celular y regional como en su regulación a través de diferentes mecanismos celulares, ha hecho muy difícil delimitar el papel de cada forma en situaciones patológicas concretas y su consideración como dianas terapéuticas novedosas.
Posiblemente el área más prometedora y donde se están realizando esfuerzos y avances en la investigación es el conocimiento de los mecanismos neuroprotectores que puedan activarse tras un episodio de isquemia cerebral, o incluso antes, en el paradigma del llamado precondicionamiento isquémico y que podrían contribuir a la recuperación funcional del cerebro dañado. Muchos de estos mecanismos conllevan procesos de plasticidad y remodelaciones tanto morfológicas como funcionales de las células gliales, y alteraciones en la neurogénesis [64].
Transportadores de glicina
La glicina, el aminoácido proteinogenético más pequeño, representa, junto con el ácido γ-aminobutírico (GABA), uno de los dos transmisores inhibidores de la neurotransmisión rápida. La glicina es especialmente abundante en zonas caudales del sistema nervioso central, como el tallo cerebral, la zona pontinocerebelosa y la médula espinal. En estas áreas se une y activa los receptores de glicina (GlyRs), causando una hiperpolarización de la membrana neuronal debido a la corriente de Cl– generada. En el tallo cerebral y la médula espinal, las interneuronas glicinérgicas inhibitorias controlan la generación de ritmos motores, la coordinación de respuestas reflejas espinales y el procesamiento de señales sensoriales y nociceptivas [65]. Las neuronas glicinérgicas inhibitorias se encuentran en numerosas zonas del sistema nervioso central, aunque son especialmente abundantes en las astas dorsales de la médula espinal, particularmente en la lámina III [66]. Las interneuronas espinales glicinérgicas del tipo Ia median circuitos reflejos de inhibición recíproca, permitiendo de esta forma la relajación de músculos antagónicos y la contracción coordinada de músculos agonistas, mientras que las interneuronas de Renshaw regulan la excitabilidad de las motoneuronas mediante la producción de señales inhibidoras recurrentes a través de un sistema de retroalimentación negativa [67,68]. Por otra parte, la glicina es un importante neurotransmisor implicado en el procesamiento de la información auditiva en los núcleos cocleares, en el complejo de la oliva superior y en el colículo inferior, donde interviene en la modulación de diversos circuitos neuronales [68,69]. Asimismo, la glicina está implicada en la supresión de las señales nociceptivas en la médula espinal [70]. Un importante aspecto de las acciones de la glicina se refiere al procesamiento de la información visual: hay neuronas glicinérgicas inhibidoras involucradas en la modulación de los campos receptivos en la retina [71].
La glicina se considera actualmente un neurotransmisor excitador en muchas regiones del sistema nervioso central al actuar como un coagonista obligado junto con el glutamato en los receptores ionotrópicos de glutamato del tipo NMDA (N-metil-D-aspartato), los cuales, tras su activación, producen una despolarización de la membrana neuronal [72,73].
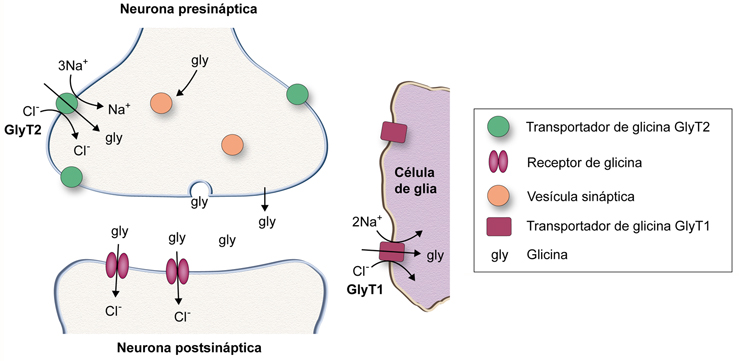
De una forma similar a la descrita para el caso del glutamato, las concentraciones de glicina extracelulares deben mantenerse en unos límites de concentración muy estrictos. Los niveles de glicina extracelulares en condiciones de equilibrio deben estar en el rango de nanomoles, mientras que sus niveles intracelulares se mantienen entre 10 y 20 mM. Las bajísimas concentraciones de glicina extracelulares se mantienen mediante el funcionamiento de transportadores de glicina situados en la membrana plasmática de las neuronas y las células de la glía, lo que permite la activación precisa y específica de los receptores de glicina. Por otra parte, los altos niveles de glicina intracelulares permiten un rellenado eficiente de las vesículas sinápticas a través de los transportadores vesiculares de aminoácidos inhibidores [74,75] (Fig. 2).
Figura 2. Esquema de una sinapsis glicinérgica inhibitoria. Se representan los transportadores de glicina GlyT1 y GlyT2, y el receptor postsináptico de glicina. La glicina almacenada en vesículas sinápticas, una vez liberada al espacio intersináptico, se une a receptores postsinápticos, abriendo el paso de iones cloruro y causando la hiperpolarización del terminal. La glicina liberada es recapturada mediante los transportadores de glicina del tipo GlyT1 (en la glía) o GlyT2 (en las neuronas glicinérgicas).
Se conocen dos tipos de transportadores de glicina GlyT1 y GlyT2. Ambas proteínas pertenecen a la familia SLC6 o NSS, que incluye los transportadores de los neurotransmisores noradrenalina, dopamina, serotonina y GABA, así como a transportadores de sustancias no neurotransmisoras, como osmolitos o aminoácidos. GlyT1 y GlyT2 están codificados por los genes SLC6A9 y SLC6A5, respectivamente [74], y son proteínas homólogas que ostentan alrededor del 50% de identidad de secuencia. Hasta la fecha se han descrito cinco variantes de GlyT1 (GlyT1a-e) y tres variantes de GlyT2 (GlyT2a-c) como resultado del uso alternativo de promotores, o como resultado de un splicing alternativo.
GlyT1 tiene una distribución mayoritariamente rostral y se concentra en los astrocitos que circundan y envuelven tanto las sinapsis inhibidoras glicinérgicas como las excitadoras glutamatérgicas, y se encuentra, además, en tejidos periféricos como el páncreas y el hígado [76–78]. GlyT2 reside principalmente en áreas caudales del sistema nervioso central, como la médula espinal, el tallo cerebral y el cerebelo, donde se halla exclusivamente localizado en los axones y terminales de las neuronas glicinérgicas [79]. Los dos transportadores de glicina no sólo difieren en su localización regional y celular, también se diferencian en sus propiedades funcionales mediante un acoplamiento termodinámico diferencial que hace que ambos transportadores posean capacidades concentrativas muy diferentes. Ambos transportadores son dependientes de los iones Na+ y Cl– para el transporte acoplado de glicina, pero mientras que GlyT1 se acopla a dos iones sodio, GlyT2 lo hace a tres iones sodio (Fig. 2). Este hecho hace que GlyT2 sea, desde el punto de vista termodinámico, mucho más eficiente que GlyT1 en cuanto al transporte concentrativo de glicina, y no sólo esto, sino que es capaz de operar en un rango mucho más amplio de concentraciones de glicina [80].
De forma similar al resto de transportadores de la familia SLC6/NSS, GlyT1 y GlyT2 son proteínas politópicas con 12 dominios transmembrana y extremos amino y carboxilo localizados en el interior celular. La estructura tridimensional de estos transportadores ha sido revelada por homología con una proteína bacteriana, el transportador de leucina de Aquifex aeolicus (LeuTAa), cuya estructura cristalina se ha establecido a una resolución de 1,65 Å [81]. Esta elevada resolución, infrecuente entre las proteínas de membrana, ha proporcionado numerosas claves para desentrañar las bases estructurales del funcionamiento de los transportadores de la familia SLC6/NSS. Más recientemente se han descrito las estructuras de los transportadores de dopamina de Drosophila melanogaster [82] y de serotonina humano [83] a muy alta resolución, ambos pertenecientes a la misma familia génica. GlyT2 es una proteína bastante más grande que GlyT1, debido a su extremo N-terminal intracelular de casi 200 aminoácidos. Esta característica es única entre los miembros de la familia SLC6 [84–86].
El funcionamiento molecular de estos transportadores sigue un esquema de apertura y cierre ya ampliamente aceptado. Exponen de forma alternante los sitios de unión a los sustratos a un lado y otro de la membrana, lo que permite captarlos en un compartimento y liberarlos en el opuesto. Para que esto suceda es necesario que las compuertas externa e interna no estén abiertas simultáneamente. El ciclo catalítico del transporte se inicia con la unión en el exterior de iones y sustrato a la conformación ‘hacia fuera’ del transportador que mantiene abierta la compuerta externa. Esta unión provoca simultáneamente el cierre de la compuerta externa y la apertura de la interna, con la adquisición de la conformación ‘hacia dentro’. La coordinación en el funcionamiento de las dos compuertas es clave, ya que si se abriesen simultáneamente, la energía contenida en el gradiente de sodio se perdería. Tras la liberación del sustrato y los iones al interior celular, la compuerta interna se cierra a la vez que la externa se abre, completando el ciclo [81]. La última etapa, el retorno del transportador vacío, es la más lenta y limitante del proceso global, y las mutaciones que bloquean el transportador en esta etapa impiden que el ciclo de transporte se complete, aunque no afectan al intercambio reversible de sustrato e iones a ambos lados de la membrana. Esto explica que el transporte sea más lento que el intercambio, en el que esta etapa de retorno no está implicada [87].
La información de la que se dispone actualmente sobre las funciones fisiológicas de los transportadores de glicina es bastante amplia y proviene del estudio de animales deficientes en cada una de estas proteínas y del desarrollo de compuestos que actúan como inhibidores de los transportadores. Ratones modificados genéticamente en los que no se encuentra GlyT1 muestran una neurotransmisión glicinérgica exacerbada debido a la sobreexposición de receptores de glicina postsinápticos como consecuencia de elevadas concentraciones de glicina en el espacio sináptico. Al no funcionar GlyT1, no hay capacidad de recuperación de glicina en el interior celular. Este hecho sugiere, por otra parte, que el otro transportador de glicina, GlyT2, no suple la carencia de GlyT1 y, por lo tanto, es este transportador en solitario el responsable de mantener las concentraciones extracelulares de glicina a niveles apropiados [88,89]. De forma contraria, ratones deficientes en GlyT2 muestran una neurotransmisión glicinérgica inhibitoria muy disminuida, debido a que las concentraciones intracelulares de glicina no se mantienen, lo que impide el rellenado eficiente de las vesículas sinápticas y su posterior liberación al espacio sináptico. Un fallo importante en la neurotransmisión glicinérgica inhibidora conduce a problemas motores y respiratorios graves y a la muerte poco después del nacimiento [90,91].
Durante los últimos años se han publicado revisiones muy completas que describen el desarrollo de compuestos que interfieren en la función de los transportadores GlyT1 y GlyT2. Nuestro grupo describió que la sarcosina (N-metilglicina) inhibe el transporte de alta afinidad de glicina en células de la glía [92,93] y más tarde se demostró que este compuesto inhibe GlyT1, pero no GlyT2 [94]. Esta es la razón por la cual la sarcosina se utilizó como ‘molde’ para el desarrollo de la primera generación de inhibidores específicos de GlyT1 [95]. La mayoría actúa como inhibidores competitivos del transportador, aunque no siempre; por ejemplo, la (±)-N-[3(4’fluo-rofenil)-3-(4’-fenilfenoxi)propil]sarcosina (NFPS) se une al transportador de forma no competitiva, con una alta afinidad y una baja velocidad de disociación, lo que hace que se eleven de forma continua los niveles de glicina en el espacio sináptico. Por este motivo, cuando la NFPS se administra a roedores, provoca una activación tónica de los receptores postsinápticos de glicina GlyRs, lo cual causa una disminución en la actividad motora y respiratoria. Varios laboratorios farmacéuticos (Merck, Pfizer, Organon, Lundbeck, Amgen) han desarrollado compuestos derivados de la NFPS con el objetivo de poder utilizarlos en situaciones como la esquizofrenia. Tanto la utilización de la NFPS y sus derivados como de otros compuestos inhibidores de GlyT1 ha sido, y sigue siendo, de gran utilidad para desvelar los mecanismos y las funciones de GlyT1 [96,97].
De forma paralela, se han desarrollado moléculas que funcionan como inhibidores de GlyT2 con varios propósitos: por un lado, investigar sobre el funcionamiento del transportador, y por otro, desarrollar compuestos que puedan ser de utilidad como analgésicos en situaciones de dolor crónico, lo cual se verá más adelante. Los dos compuestos más ampliamente utilizados son ALX1393 (Alexis, labs) y ORG25543 (Organon, labs). Ambos actúan eliminando parcialmente la actividad de GlyT2. La utilización de ambos compuestos en animales da como resultado un incremento en la neurotransmisión glicinérgica. ALX1393 tiene unos claros efectos analgésicos con relativamente pocos efectos secundarios. ORG25543 difiere con ALX1393 en su mecanismo de acción sobre GlyT2. Se comporta como un inhibidor irreversible del transportador, lo que conlleva que su uso prolongado tenga más efectos secundarios indeseados que ALX1393 [96,98].
GlyT1 y GlyT2 son proteínas sometidas a un estricto control regulador. GlyT1 se inhibe por el ácido araquidónico, un segundo mensajero liberado tras la activación de la fosfolipasa A2 [99,100]. Además, se modula por niveles de pH y por concentraciones de Zn2+ [101,102]. La regulación de la actividad de GlyT2 ha resultado ser compleja, al estar involucrados diferentes mecanismos en el control del tráfico intracelular. Cabe destacar la activación de la PKC [103–105], los receptores purinérgicos P2X y P2Y [106], la chaperona calnexina [107], la modificación postraduccional de ubiquitinación [108] y la interacción de GlyT2 con diferentes proteínas: Na+/K+-ATPasa [109], Ca2+-ATPasa [110], el intercambiador Na+/Ca2+ y la proteína SNARE sintaxina-1a [111], así como por las ‘enzimas dependientes de Ca2+/calmodulina’ [112]. Por otra parte, recientemente se ha establecido la asociación de GlyT2 con microdominios lipídicos ricos en colesterol y fosfolípidos, lo que ha puesto de manifiesto un estadio de complejidad superior [113]. La modificación farmacológica de los componentes lipídicos de los dominios reduce tanto su inclusión en ellos como su actividad de transporte de glicina, lo que indica que GlyT2 requiere la localización en balsas lipídicas para mantener una función óptima [114]. Además, la endocitosis del transportador y su reciclaje a la membrana podrían estar relacionados con su asociación a dominios concretos de la membrana plasmática [115].
Implicación de los transportadores de glicina en estados patológicos
Dolor neuropático
En condiciones fisiológicas normales, cualquier proceso de lesión en tejidos periféricos o un proceso inflamatorio llevan aparejada la activación de neuronas nociceptivas especializadas en la transmisión de dolor. Este fenómeno es un mecanismo de alerta, agudo, reversible y conlleva cambios adaptativos del sistema. Sin embargo, un daño en el sistema nervioso central o periférico puede conducir a la aparición de un dolor crónico persistente más allá del tiempo en el que se ha producido la lesión con una duración de entre tres a seis meses y que se caracteriza por dolor espontáneo, hiperalgesia y alodinia, tanto con estímulos térmicos como mecánicos. Desde hace tiempo se conoce la relación existente entre situaciones de dolor crónico y la adaptación patológica persistente que se produce en la neurotransmisión excitadora, inhibidora y en las relaciones neurona-glía.
La información de dolor se transmite desde la periferia hasta el tálamo a través de centros espinales que utilizan, entre otros, mecanismos glutamatérgicos. En este sistema, una serie de interneuronas glicinérgicas y gabérgicas filtran y modulan el flujo de información, de forma que un desequilibrio entre sistemas de neurotransmisión excitadora e inhibidora en la médula espinal conduce a un incremento de las respuestas a estímulos nocivos. Una reducción en la neurotransmisión inhibitoria, así como un aumento en la neurotransmisión excitadora en la médula espinal, contribuye al desarrollo de un incremento en la sensibilidad al dolor. Como se indicaba al comienzo de este apartado, las neuronas y los receptores glicinérgicos inhibidores son especialmente abundantes en las astas dorsales de la médula espinal, particularmente en la lámina III, coincidiendo con las regiones donde hay una especial abundancia de transportadores GlyT1 y GlyT2 y de receptores de glicina GlyR sensibles a la estricnina [66,67,116]. Estas neuronas glicinérgicas contribuyen a la inhibición de señales nociceptivas y desempeñan un papel clave para discriminar información nociceptiva de información no nociva (Fig. 3). La desregulación de estos sistemas glicinérgicos, junto con los gabérgicos, subyace al dolor neuropático o inflamatorio [117]. Además de las vías de transmisión de dolor antes descritas, existe un sistema descendente de inervación glicinérgica importante en la médula espinal que arranca en áreas supraespinales [116]. La estimulación de estas vías inhibitorias descendentes inhibe la nocicepción. Se trata de un número importante de neuronas glicinérgicas y gabérgicas localizadas en la zona rostral ventromedial de la médula que proyectan a la lámina I-II y a la IV-V [118]. Todas estas vías descendentes parece ser que se encuentran inhibidas o abolidas en el dolor crónico y pueden contribuir a la sintomatología de esta patología [119,120].
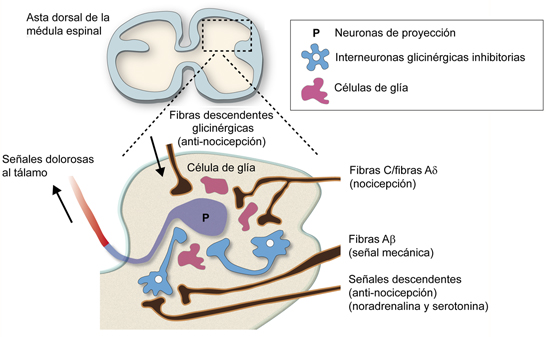
Figura 3. Esquema de las vías glicinérgicas de la médula espinal implicadas en el procesamiento del dolor. Las neuronas de proyección (P) localizadas en capas superficiales de las astas dorsales de la médula espinal que se proyectan hacia el tálamo están estrechamente reguladas por señales glicinérgicas inhibitorias. La inhibición de los transportadores de glicina GlyT1 y GlyT2 conduce al aumento de los niveles de glicina en el espacio intersináptico, provocando la activación de la neurotransmisión glicinérgica inhibitoria. Las fibras nociceptivas del tipo C y Ab estimulan las neuronas de proyección (P) y activan las interneuronas glicinérgicas (que median las señales antinociceptivas noradrenérgicas y serotoninérgicas descendentes), las cuales inhiben las neuronas de proyección dolorosa (figura modificada, con permiso de [90] y [130]).
Esquizofrenia
La esquizofrenia es una enfermedad compleja que afecta a alrededor del 1% de la población mundial y constituye una de las causas más importantes de discapacidad crónica. Aunque su etiología se desconoce, la enfermedad implica diversas anomalías neuromorfológicas y neuroquímicas, y se acepta que factores genéticos, bien solos o potenciados por factores ambientales y epigenéticos, desempeñan un papel importante en su patogenia. Numerosos estudios realizados durante los últimos 40 años han relacionado la patología de las psicosis esquizofrénicas con alteraciones en la neurotransmisión mediada por aminas biógenas, la neurotransmisión glutamatérgica y la gabérgica. Aunque desde hace bastantes años la esquizofrenia se ha tratado con antipsicóticos antagonistas de receptores dopaminérgicos del tipo D2, estudios preclínicos y clínicos realizados en la última década han llevado a desarrollar la hipótesis glutamatérgica, que postula que muchos de los síntomas de la patología que afectan al deterioro cognitivo se deben a una reducida función del receptor de glutamato del tipo NMDA. Como se describió al inicio de este apartado, el transportador de glicina GlyT1 desempeña un papel doble, regulando los niveles de glicina en la sinapsis inhibidora glicinérgica y excitadora glutamatérgica, lo que convierte a esta proteína en una diana terapéutica potencial para algunas patologías asociadas a alteraciones de estas vías nerviosas. Puesto que GlyT1 regula las concentraciones de glicina en el microentorno de los receptores de NMDA y la reducción de la actividad de GlyT1 neuronal mejora el aprendizaje asociativo, en la actualidad parece claro que el bloqueo de transportador GlyT1 constituye un abordaje farmacológico para el tratamiento de la esquizofrenia [96].
Hiperplexia hereditaria
La hiperplexia hereditaria o enfermedad del sobresalto (denominada en ocasiones ‘síndrome del bebé entumecido’, OMIM 149400) es un síndrome clínico poco común que se manifiesta muy pronto después del nacimiento o incluso en el período intrauterino, caracterizado por sobresaltos enérgicos, exagerados y sostenidos en respuesta a estímulos triviales inesperados auditivos, somatosensoriales o visuales [121,122]. La reacción tras estímulos de este tipo es una apreciable rigidez del tronco y las extremidades, puños apretados y frecuentes temblores, que en ocasiones pueden recordar actitudes o respuestas propias de ataques epilépticos tónicos, lo que puede llevar a un diagnóstico erróneo de daño cerebral perinatal con espasticidad y síntomas epilépticos. Sin embargo, el electroencefalograma es normal y existen una serie de tests motores que ayudan a discriminar síntomas y permiten llegar a un diagnóstico claro [123]. Aunque con el paso de los meses alguno de los síntomas iniciales, como la rigidez muscular, pueda atenuarse, el riesgo de muerte súbita del bebé es alto como consecuencia de fallos cardiorrespiratorios y espasmos laríngeos. Con un desarrollo mental generalmente normal, en los niños con hiperplexia hereditaria subsisten alteraciones motoras a lo largo de la vida, más claramente hasta la adolescencia, como los espasmos exagerados ante estímulos externos triviales. Los individuos afectados se mantienen erguidos con dificultad, caminan con una gran inseguridad y requieren, en muchos casos, asistencia externa. Esta sintomatología se hace especialmente patente cuando se enfrentan a un apuro, en una concentración de gente o si se ven, de algún modo, forzados por la prisa. Como consecuencia de estas situaciones, no son infrecuentes caídas fortuitas, con el consiguiente riesgo de lesiones importantes.
Se han descrito varias mutaciones en el gen SLC6A5, localizado en el brazo corto del cromosoma 11, que codifica el transportador presináptico de glicina, y que causan hiperplexia. El gen posee 16 exones distribuidos en una región de aproximadamente 21,4 Mb, en la que sólo el primer exón contiene el sitio de iniciación de la traducción. De los 16 exones del gen SLC6A5, en varios de ellos se han encontrado mutaciones relacionadas con hiperplexia [124,125]. La mayor parte tiene carácter recesivo, aunque en un caso era dominante. Algunas de estas mutaciones generan un transportador truncado y, por tanto, inactivo. Otras son mutaciones puntuales o sinsentido que, aunque producen una proteína que se expresa en la membrana plasmática, es una proteína inactiva por fallos en la unión de alguno de sus ligandos glicina o Na+, o por estar la sustitución en regiones cruciales para la actividad de transporte. En otros polimorfismos de hiperplexia hereditaria se produce un GlyT2 que queda retenido en compartimentos intracelulares, impidiendo su llegada a la membrana plasmática [126].
Hiperglicinemia no cetósica
La hiperglicinemia no cetósica (OMIM 605899) es una enfermedad rara autosómica recesiva muy poco frecuente que cursa con una encefalopatía grave causada por una deficiencia en el sistema mitocondrial de ruptura de la glicina [127]. Aunque existen diferentes fenotipos entre los pacientes, la mayoría se caracteriza por hipotonía, convulsiones, problemas cognitivos, retraso en el desarrollo y espasmos mioclónicos que conducen a períodos de apnea e incluso a la muerte. Pacientes con este tipo de hiperglicinemia no cetósica, muy grave desde el punto de vista clínico, se caracterizan por presentar unos niveles de glicina extraordinariamente altos en el plasma y en el líquido cefalorraquídeo. Sin embargo, recientemente se ha descrito un fenotipo más benigno de la enfermedad en la que los individuos afectados presentan niveles moderadamente elevados de glicina en el líquido cefalorraquídeo y niveles normales en el suero. Estos pacientes no poseen mutaciones en el sistema mitocondrial de ruptura de glicina, sino mutaciones en el gen SLC6A9, que codifica el transportador de glicina GlyT1 [128–130].
Transportadores de glicina como potenciales dianas terapéuticas
En el tratamiento sintomático de la esquizofrenia se han venido utilizando antipsicóticos de primera y segunda generación, casi todos dirigidos contra receptores de dopamina de los tipos D1 y D2, así como receptores de serotonina. Los efectos secundarios (sedación, ganancia de peso, disfunción sexual, etc.) de estos fármacos se basan en su baja especificidad; por otra parte, sólo son efectivos en una parte de pacientes y alivian sólo algunos síntomas. El convencimiento más reciente de que en la esquizofrenia se produce un déficit en el funcionamiento de los receptores glutamatérgicos NMDA y su relación con el transportador de glicina GlyT1 ha despertado un gran interés en el desarrollo de inhibidores de este transportador como nuevos agentes antipsicóticos [131,132]. La glicina es un agonista obligado del receptor NMDA; si de alguna forma podemos manipular los niveles de glicina en el microentorno de los receptores NMDA, provocaremos su estimulación. La manera más directa de conseguir esto es interfiriendo en la recaptura de glicina en el interior de las neuronas y de las células de la glía mediante inhibidores del transportador de glicina GlyT1. Estos compuestos son efectivos en el alivio de la mayor parte de los síntomas típicos de la esquizofrenia (síntomas positivos, síntomas negativos y deterioro cognitivo) (Fig. 4).
Durante los últimos años, diversas compañías farmacéuticas han desarrollado inhibidores de GlyT1 pertenecientes a varias familias de compuestos con modalidades de actuación diferentes y afinidades diferentes. Muchos de estos compuestos se encuentran en fases clínicas avanzadas y abren posibilidades muy prometedoras para el tratamiento no sólo de la esquizofrenia, sino de otras enfermedades como la adicción al alcohol, la ansiedad y algunos tipos de depresión, en las cuales, de una u otra forma, se encuentran involucrados los receptores de glutamato NMDA [96]. De entre todos los inhibidores de GlyT1 conocidos hasta la fecha, la bitopertina (conocida también como RG1678 y RO4917838) es el compuesto que se encuentra en fases clínicas de desarrollo más avanzadas (actualmente en fase clínica III). La bitopertina es el primer compuesto que representa de forma más clara la aplicación directa de la hipótesis glutamatérgica a la práctica clínica, y puede ser el inicio de una nueva clase de antipsicótico de tercera generación [96] (Fig. 4).
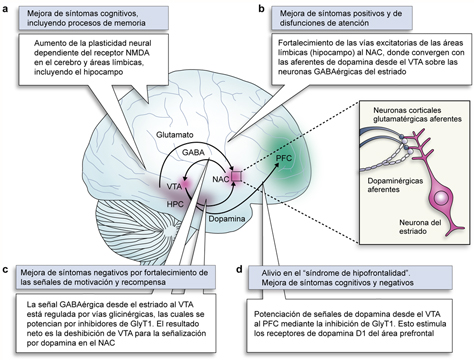
Figura 4. Se representan los posibles efectos antipsicóticos resultantes de la inhibición del transportador de glicina GlyT1. HPC, hipocampo; NAC, núcleo acumbens; PFC, corteza prefrontal; VTA, área tegmental central. Tras la inhibición del transportador de glicina GlyT1 se espera observar los siguientes efectos: a) una potenciación de los receptores de glutamato del tipo NMDA que debería afectar positivamente a funciones corticales, incluyendo áreas de la corteza asociativa y del sistema límbico; b) un aumento en las señales desde el sistema límbico hacia el núcleo accumbens propiciado por la activación de los receptores NMDA; c) la inhibición del transportador de glicina GlyT1 podría activar señales de recompensa mediadas por conexiones neuronales ente el VTA y el NAC debido a la desinhibición de señales gabérgicas desde el NAC al VTA (este hecho podría tener un efecto positivo sobre los síntomas negativos característicos de esquizofrenia, como la apatía o la falta de motivación); y d) la desinhibición del VTA podría reforzar vías dopaminérgicas a la corteza prefrontal influyendo positivamente en el llamado ‘síndrome de hipofrontalidad’ característico de la enfermedad, que es responsable de síntomas cognitivos y negativos de la esquizofrenia, incluyendo disfunciones en la memoria de trabajo, las funciones ejecutivas, el comportamiento y la atención (figura modificada, con permiso de [90]).
Por otra parte, recientemente se ha demostrado en modelos preclínicos de dolor que la inhibición de GlyT1 o de GlyT2 puede ser un método muy efectivo de mejorar el dolor neuropático con muchos menos efectos secundarios que los típicos de los tratamientos actuales con antidepresivos tricíclicos [133–135]. Era conocida la implicación de la neurotransmisión glicinérgica en los mecanismos de dolor neuropático: la inhibición de los receptores de glicina GlyR en la médula espinal por estricnina induce hiperalgesia y alodinia mecánica y, por otra parte, el aumento en la neurotransmisión glicinérgica mediante la inhibición de los transportadores de glicina reduce el dolor [135–137] (Fig. 5).
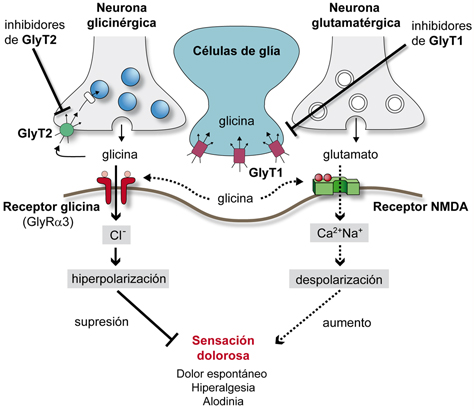
Figura 5. Localización y funciones de los transportadores de glicina en las sinapsis inhibitorias y excitatorias. Supresión de señales dolorosas como consecuencia de la inhibición de los transportadores de glicina GlyT1 y GlyT2. En las sinapsis excitatorias glutamatérgicas, la glicina actúa como un agonista obligado del receptor de glutamato del tipo NMDA. La activación de este receptor lleva a una apertura del canal y a la entrada de cationes al terminal postsináptico con la consecuente despolarización del terminal. La neurotransmisión es regulada mediante la recaptura de glicina a través de sus transportadores dependientes de iones sodio y cloruro. GlyT2 está localizado en los terminales presinápticos de neuronas glicinérgicas y se encarga de mantener niveles de glicina intracelulares adecuados para asegurar el relleno de glicina a las vesículas sinápticas, las cuales aseguran la liberación del neurotransmisor para su interacción con receptores postsinápticos de glicina. GlyT1 se expresa principalmente en las células de la glía y, en menor medida, en los terminales presinápticos glutamatérgicos. Este transportador es el responsable de mantener niveles de glicina en el espacio extracelular adecuados para actuar sobre los receptores de glutamato del tipo NMDA como un agonista obligado. La inhibición de GlyT1 llevaría a un aumento de la concentración de glicina en los receptores de glicina GlyR, una estimulación de las vías inhibitorias glicinérgicas y una supresión de la sensación dolorosa. Por otra parte, el incremento de glicina en las proximidades de los receptores NMDA llevaría a su activación y, como consecuencia, a un aumento en la sensación dolorosa y al desarrollo de efectos psicóticos (figura modificada, con permiso de [90] y [130]).
Bibliografía
C. Giménez, F. Zafra, C. Aragón[REV NEUROL 2018;67:491-
↵ 1. Butcher SP, Hamberger H. In vivo studies on the extracellular, and veratrine-releasable, pools of endogenous amino acids in the rat striatum: effects of corticostriatal deafferentation and kainic acid lesion. J Neurochem 1987; 48: 713-21.
↵ 2. Danbolt NC. Glutamate uptake. Prog Neurobiol 2001; 65: 1-105.
↵ 3. Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. J Neurochem 1987; 7: 357-68.
↵ 4. Choi DW. Excitotoxic cell death. J Neurobiol 1992; 23: 1261-76.
↵ 5. Tzingounis AV, Wadiche JL. Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat Rev Neurosci 2007; 8: 935-47.
↵ 6. Gegelashvili G, Schousboe A. High affinity glutamate transporters: regulation of expression and activity. Mol Pharmacol 1997; 52: 6-15.
↵ 7. Seal RP, Amara SG. Excitatory amino acid transporters: a family in flux. Annu Rev Pharmacol Toxicol 1999; 39: 431-56.
↵ 8. Vandenberg RJ, Ryan RM. Mechanisms of glutamate transport. Physiol Rev 2013; 93: 1621-57.
↵ 9. Utsunomiya-Tate N, Endou H, Kanai Y. Tissue specific variants of glutamate transporter GLT-1. FEBS Lett 1997; 416: 312-6.
↵ 10. Peacey E, Miller CC, Dunlop J, Rattray M. The four major N- and C-terminal splice variants of the excitatory amino acids transporter GLT-1 form cell surface homomeric and heteromeric assemblies. Mol Pharmacol 2009; 75: 1062-73.
↵ 11. Cavellier P, Attwell D. Tonic release of glutamate by DIDS-sensitive mechanism in rat hippocampal slices. J Physiol 2005; 564: 397-410.
↵ 12. Herman MA, Jahr CE. Extracellular glutamate concentration in hippocampal slice. J Neurosci 2007; 27: 9736-41.
↵ 13. Zerange N, Kavanaugh MP. Flux coupling in a neuronal glutamate transporter. Nature 1996; 383: 634-7.
↵ 14. El Mestikawy S, Wallén-Mackenzie M, Fortin GM, Descarries L, Trudeau LE. From glutamate co-release to vesicular synergy: vesicular glutamate transporters. Nat Rev Neurosci 2011; 12: 204-16.
↵ 15. Lewy LM, Warr O, Attwell D. Stoichiometry of the glial glutamate transporter GLT-1 expressed inducibly in a Chinese hamster ovary cell line selected for low endogenous N+-dependent glutamate uptake. J Neurosci 1998; 18: 9620-8.
↵ 16. Owe SG, Marcaggi P, Attwell D. The ionic stoichiometry of the GAST glutamate transporter in salamander retinal glia.
J Physiol 2006; 577: 591-9.
↵ 17. Billups B, Rossi D, Attwell D. Anion conductance behavior of the glutamate uptake carrier in salamander retinal glial cells. J Neurosci 1996; 16: 6722-31.
↵ 18. Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 1995; 375: 599-603.
↵ 19. Vandenberg RJ, Arriza JL, Amara SG, Kavanaugh MP. Constitutive ion fluxes and substrate binding domains of human glutamate transporters. J Biol Chem 1995; 270: 17668-71.
↵ 20. Dehnes Y, Chaudry FA, Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC. The glutamate transporter EAAT4 in rat cerebellar Purkinje cells: a glutamate-gated chloride channel concentrated near the synapse in parts of the dendritic membrane facing astroglia. J Neurosci 1998; 18: 3606-10.
↵ 21. Furuta A, Martin LJ, Lin, CL, Dykes-Hoberg M, Rothstein JD. Cellular and synaptic localization of the neuronal glutamate transporters EAAT3 and EAAT4. Neuroscience 1997; 81: 1031-42.
↵ 22. Tanaka J, Ichikawa R, Watanabe T, Tanaka K, Inoue Y. Extra-junctional localization of glutamate transporter EAAT4 at excitatory Purkinje cell synapses. NeuroReport 1997; 8: 2461-4.
↵ 23. Tanaka K. Functions of glutamate transporters in the brain. Neurosci Res 2000; 37: 15-9.
↵ 24. Pow DV, Barnett NL. Developmental expression of excitatory amino acid transporter 5: a photoreceptor and bipolar cell glutamate transporter in rat retina. Neurosci Lett 2000; 280: 21-4.
↵ 25. Schneider N, Cordeiro S, Machtens JP, Braams S, Rauen T, Fahlke C. Functional properties of the retinal glutamate transporter GLT-1c and EAAT5. J Biol Chem 2014; 289: 1815-24.
↵ 26. Danbolt NC, Storm-Matisen J, Kanner BI. A Na and K coupled L-glutamate transporter purified from rat brain is located in glial cell processes. Neuroscience 1992; 51: 295-310.
↵ 27. Rothstein JD, Martin L, Lvey AL, Dykes-Hoberg M, Jin L, Wu D, et al. Localization of neuronal and glial glutamate transporters. Neuron 1994; 13: 713-25.
↵ 28. Lehre KP, Levy LM, Ottersen OP, Storm-Mathissen J, Danbolt NC. Differential expression of two glial glutamate transporters in rat brain: quantitative and immunocytochemical observations. J Neurosci 1995; 15: 1835-53.
↵ 29. Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 1997; 276: 1699-702.
↵ 30. Petr GT, Sun Y, Frederick NM, Zhou Y, Dhamne SC, Mustafa Q, et al. Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J Neurosci 2015; 35: 5187-201.
↵ 31. Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, et al. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci 1998; 10: 976-88.
↵ 32. Yemool D, Boudker O, Jin Y, Gouaux E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 2004; 431: 811-8.
↵ 33. Nothmann D, Leinenweber A, Torres-Salazar D, Kovermann P, Hotzy J, Gameiro A, et al. Hetero-oligomerization of neuronal glutamate transporters. J Biol Chem 2011; 286: 3935-43.
↵ 34. Grewer C, Balani P, Weidenfeller P, Bartusell T, Tao Z, Rauen T. Individual subunits of the glutamate transporter EAAC1 homotrimer function independently of each other. Biochemistry 2005; 44: 11913-23.
↵ 35. Boudker O, Ryan RM, Yernool D, Shimamoto K, Gouaux E. Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature 2007; 445: 387-93.
↵ 36. Seal RP, Amara SG. A reentrant loop domain in the glutamate carrier EAAT1 participates in substrate binding and translocation. Neuron 1998; 21: 1487-98.
↵ 37. Slotboom DJ, Lolkema JS, Konnings WN. Membrane topology of the C-terminal half of the neuronal, glial, and bacterial glutamate transporter family. J Biol Chem 1996; 71: 31317-21.
↵ 38. Ruan Y, Miyaqi A, Wang X, Chami M, Boudker O, Scheuring S. Direct visualization of glutamate transporter elevator mechanism by high-speed AFM. Proc Natl Acad Sci USA 2017; 114: 1584-8.
↵ 39. Casado M, Bendahan A, Zafra F, Danbolt NC, Aragón C, Giménez C, et al. Phosphorylation and modulation of brain glutamate transporters by protein kinase C. J Biol Chem 1993; 268: 27313-7.
↵ 40. Ganel R, Crosson CE. Modulation of human glutamate transporter activity by phorbol ester. J Neurochem 1998; 70: 993-1000.
↵ 41. Kalandadze A, Wu Y, Robinson MB. Protein kinase C activation decreases cell surface expression of the GLT-1 subtype of glutamate transporter. Requirement of a carboxyl-terminal domain and partial dependence on serine 486. J Biol Chem 2002; 277: 45741-50.
↵ 42. Murphy-Royal C, Dupuis JP, Varela JA, Panatier A, Pinson B, Baufreton J, et al. Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nat Neurosci 2015; 18: 219-26.
↵ 43. García-Tardón N, González-González IM, Martinez-Villarreal J, Fernández-Sánchez E, Giménez C, Zafra F. Protein kinase C (PKC)-promoted endocytosis of glutamate transporter GLT-1 requires ubiquitin ligase Nedd4-2-dependent ubiquitination but not phosphorylation. J Biol Chem 2012; 287: 19177-87.
↵ 44. Martínez-Villarreal J, García-Tardón N, Ibáñez I, Giménez C, Zafra F. Cell surface turnover of the glutamate transporter GLT-1 is mediated by ubiquitination/deubiquitination. Glia 2012; 60: 1356-65.
↵ 45. Ibáñez I, Díez-Guerra FJ, Giménez C, Zafra F. Activity dependent internalization of the glutamate transporter GLT-1 mediated by β-arrestin 1 and ubiquitination. Neuropharmacology 2016; 107: 376-86.
↵ 46. Zafra F, Ibáñez I, Giménez C. Glutamate transporters: the arrestin connection. Oncotarget 2017; 8: 5664-5.
↵ 47. Jiménez E, Núñez E, Ibáñez I, Draffin JE, Zafra F, Giménez C. Differential regulation of the glutamate transporters GLT-1 and GLAST by GSK3β. Neurochem Int 2014; 79: 33-43.
↵ 48. Mitani A, Tanaka K. Functional changes of glial glutamate transporter GLT-1 during ischemia: an in vivo study in the hippocampal CA1 of normal mice and mutant mice lacking GLT-1. J Neurosci 2003; 23: 7176-82.
↵ 49. Dirgani U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 1999; 22: 391-7.
↵ 50. Stelmashook EV, Weih M, Zorov D, Victorov I, Dirnagl U, Isaev N. Short-term block of Na+/K+-ATPase in neuro-glial cell cultures of cerebellum induces glutamate dependent damage of granule cells. FEBS Lett 1999; 456: 41-4.
↵ 51. Lo EH, Moskowitz MA, Jacobs TP. Exciting, radical, suicidal: how brain cells die after stroke. Stroke 2005; 36: 189-92.
↵ 52. Longuemare MC, Swanson RA. Excitatory amino acids release from astrocytes during energy failure by reversal of sodium-dependent uptake. J Neurosci Res 1995; 40: 379-86.
↵ 53. Rothstein JD. Excitotoxic mechanisms in the pathogenesis of amyotrophic lateral sclerosis. Adv Neurol 1995; 68: 7-27.
↵ 54. Bristol LA, Rothstein JD. Glutamate transporter gene expression in amyotrophic lateral sclerosis motor cortex. Ann Neurol 1996; 39: 676-9.
↵ 55. Brujin LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997; 8: 327-38.
↵ 56. Jacob CP, Koutsilieri E, Bartl J, Neuen-Jacob E, Arzberger T, Zander N. et al. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J Alzheimer Dis 2007; 11: 97-116.
↵ 57. Li S, Mallory M, Alford M, Tanaka S, Masliah E. Glutamate transporter alterations in Alzheimer disease are possibly associated with abnormal APP expression. J Neuropathol Exp Neurol 1997; 56: 901-11.
↵ 58. Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med 2000; 6: 67-70.
↵ 59. Werner P, Pitt D, Raine CS. Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol 2001; 50: 169-80.
↵ 60. Ohgoh M, Hanada T, Smith T, Hashimoto T, Ueno M, Yamanishi Y, et al. Altered expression of glutamate transporters in experimental autoimmune encephalomyelitis. J Neuroimmunol 2002; 125: 170-8.
↵ 61. Smith T, Groom A, Zhu B. Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med 2000; 6: 62-6.
↵ 62. Matute C, Domercq M, Fogarty DJ, Pascual de Zulueta M, Sánchez-Gómez MV. On how altered glutamate homeostasis may contribute to demyelinating diseases of the CNS. Adv Exp Med Biol 1999; 468: 97-107.
↵ 63. Matute C, Alberdi E, Domercq M, Pérez-Cerdá F, Pérez-Samartín A, Sánchez-Gómez MV. The link between excitotoxic oligodendroglial death and demyelinating diseases. Trends Neurosci 2001; 24: 224-30.
↵ 64. Ji YF, Zhou L, Xie YJ, Xu SM, Zhu J, Teng P, et al. Upregulation of glutamate transporter GLT-1 by mTOR-Akt-NF-кB cascade in astrocytic oxygen-glucose deprivation. Glia 2013; 6: 1959-75.
↵ 65. Ottersen OP, Storm-Mathisen J. Cellular and subcellular localization of glycine studied by quantitative microscopic immunocytochemistry. In Ottersen OP, Storm-Mathisen J, eds. Glycine neurotransmission. Chichester, UK: John Wiley; 1990. p. 303-28.
↵ 66. Hossaini M, French PJ, Holstege JC. Distribution of glycinergic neuronal somata in the rat spinal cord. Brain Res 2007; 1142: 61-9.
↵ 67. Legendre P. The glycinergic inhibitory synapse. Cell Mol Life 2001; 58: 760-93.
↵ 68. Zafra F, Aragón C, Giménez C. Molecular biology of glycinergic neurotransmission. Mol Neurobiol 1997; 14: 117-42.
↵ 69. Wenthold R, Hunter C. Immunochemistry of glycine and glycine receptors in the central auditory system. In Ottersen OP, Storm-Mathisen J, eds. Glycine neurotransmission. Chichester, UK: John Wiley; 1990. p. 391-415.
↵ 70. Harvey RJ, Depner UB, Wässle H, Ahmadi S, Heindl C, Reinold H, et al. GlyR alpha 3: an essential target for spinal PGE(2)-mediated inflammatory pain sensitization. Science 2004; 304: 884-7.
↵ 71. Pourcho RG, Goebel DJ. Autoradiographic and immunocyto-chemical studies of glycine containing neurons in the retina. In Ottersen OP, Storm-Mathisen J, eds. Glycine neuro-transmission. Chichester, UK: John Wiley; 1990. p. 355-89.
↵ 72. Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 1987; 325: 529-31.
↵ 73. Wolosker H. NMDA receptor regulation by D-serine new findings and perspectives. Mol Neurobiol 2007; 36: 152-64.
↵ 74. Aragón C, López-Corcuera B. Glycine transporters: crucial roles of pharmacological interest revealed by gene deletion. Trends Pharmacol Sci 2005; 26: 283-6.
↵ 75. Eulenburg V, Armsen W, Betz H, Gomeza J. Glycine transporters: essential regulators of neurotransmission. Trends Biochem Sci 2005; 30: 325-33.
↵ 76. Zafra F, Aragón C, Olivares L, Danbolt NC, Giménez C, Storm-Mathisen J. Glycine transporters are differentially expressed among CNS cells. J Neurosci 1995; 15: 3952-69.
↵ 77. Zafra F, Gomeza J, Olivares L, Aragón C, Giménez C. Regional distribution and developmental variation of the glycine transporters GLYT1 and GLYT2 in the rat CNS. Eur J Neurosci 1995; 7: 1342-52.
↵ 78. Cubelos B, Giménez C, Zafra F. Localization of the GLYT1 glycine transporter at glutamatergic synapses in the rat brain. Cereb Cortex 2005; 15: 448-59.
↵ 79. Poyatos I, Ponce J, Aragón C, Giménez C, Zafra F. The glycine transporter GLYT2 is a reliable marker for glycine-immuno-reactive neurons. Brain Res Mol Brain Res 1997; 49: 63-70.
↵ 80. Roux MJ, Supplisson S. Neuronal and glial glycine transporters have different stoichiometries. Neuron 2000; 25: 373-83.
↵ 81. Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl–-dependent neurotransmitter transporters. Nature 2005; 437: 215-23.
↵ 82. Penmatsa A, Wang KH, Gouaux E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 2013; 503: 85-90.
↵ 83. Coleman JA, Green EM, Gouaux E. X-ray structures and mechanism of the human serotonin transporter. Nature 2016; 532: 334-39.
↵ 84. Bala PA, Foster J, Carvelli L, Henry LK. SLC6 transporters: structure, function, regulation, disease association and therapeutics. Mol Aspects Med 2013; 34: 197-219.
↵ 85. Anders S, Kristensen JA, Jørgensen TN, Sørensen L, Eriksen J, Loland CJ, et al. SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol Rev 2011; 63: 585-640.
↵ 86. Rudnick G, Krämer R, Blakely RD, Murphy DL, Verrey F. The SLC6 transporters: perspectives on structure, functions, regulation, and models for transporter dysfunction. Pflugers Arch 2014; 466: 25-42.
↵ 87. Rudnick G. How do transporters couple solute movements? Mol Mem Biol 2013; 30: 355-9.
↵ 88. Gomeza J, Hülsmann S, Ohno K, Eulenburg V, Szöke K, Richter D, et al. Inactivation of the glycine transporter 1 gene discloses vital role of glial glycine uptake in glycinergic inhibition. Neuron 2003; 40: 785-96.
↵ 89. Eulenburg V, Becker K, Gomeza J, Schmitt B, Becker CM, Betz H. Mutations within the human GLYT2 (SLC6A5) gene associated with hyperekplexia. Biochem Biophys Res Commun 2006; 348: 400-5.
↵ 90. Gomeza J, Ohno K, Hülsmann S, Armsen W, Eulenburg V, Richter, DW, et al. Deletion of the mouse glycine transporter 2 results in a hyperekplexia phenotype and postnatal lethality. Neuron 2003; 40: 797-806.
↵ 91. Rousseau F, Aubrey KR, Supplisson S. The glycine transporter GlyT2 controls the dynamics of synaptic vesicle refilling in inhibitory spinal cord neurons. J Neurosci 2008; 28: 9755-68.
↵ 92. Zafra F, Giménez C. Characteristics and adaptive regulation of glycine transport in cultured glial cells. Biochem J 1989; 258: 403-8.
↵ 93. López-Corcuera B, Vázquez J, Aragón C. Purification of the sodium- and chloride-coupled glycine transporter from central nervous system. J Biol Chem 1991; 266: 24809-14.
↵ 94. Liu QR, Nelson H, Mandiyan S, López-Corcuera B, Nelson N. Cloning and expression of a glycine transporter from mouse brain. FEBS Lett 1992; 305: 110-4.
↵ 95. Singer P, Dubroqua S, Yee BK. Inhibition of glycine transporter 1: the yellow brick road to new schizophrenia therapy. Curr Pharm Des 2015; 21: 3771-87.
↵ 96. Harvey RJ, Yee BK. Glycine transporters as novel therapeutic targets in schizophrenia, alcohol dependence and pain. Nat Rev Drug Disc 2013; 12: 866-85.
↵ 97. Cioffi CL. Guzzo PR. Inhibitors of glycine transporter-1: potential therapeutics for the treatment of CNS disorders. Curr Top Med Chem 2016; 16: 3404-37.
↵ 98. Kristensen AS, Andersen J, Jørgensen TN, Sørensen L. Eriksen J, Loland CJ, et al. SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol Rev 2011; 63: 585-640.
↵ 99. Zafra F, Alcántara R, Gomeza J, Aragón C, Giménez C. Arachidonic acid inhibits glycine transport in cultured glial cells. Biochem J 1990; 271: 237-42.
↵ 100. Pearlman RJ, Aubrey KR, Vandenber, RJ. Arachidonic acid and anandamide have opposite modulatory actions at the glycine transporter, GLYT1a. J Neurochem 2003; 84: 592-601.
↵ 101. Laube B. Potentiation of inhibitory glycinergic neurotransmission by Zn2+. A synergistic interplay between presynaptic P2X2 and postsynaptic glycine receptors. Eur J Neurosci 2002; 16: 1025-36.
↵ 102. Ju P, Aubrey KR, Vandenberg RJ. Zn2+ inhibits glycine transport by glycine transporter subtype 1b. J Biol Chem 2004; 279: 22983-91.
↵ 103. Gomeza J, Zafra F, Olivares L, Giménez C, Aragón C. Regulation by phorbol esters of the glycine transporter (GLYT1) in glioblastoma cells. Biochim Biophys Acta 1995; 1233: 41-6.
↵ 104. Sato K, Adams R, Betz H, Schloss P. Modulation of a recombinant glycine transporter (GLYT1b) by activation of protein kinase C. J Neurochem 1995, 65: 1967-73.
↵ 105. Fernández-Sánchez E, Martínez-Villarreal J, Giménez C, Zafra F. Constitutive and regulated endocytosis of the glycine transporter GLYT1b is controlled by ubiquitination. J Biol Chem 2009; 284: 19482-92.
↵ 106. Jiménez E, Zafra F, Pérez-Sen R, Delicado EG, Miras-Portugal MT, Aragón C, et al. P2Y purinergic regulation of the glycine neurotransmitter transporters. J Biol Chem 2011; 286: 10712-4.
↵ 107. Arribas-González E, Alonso-Torres P, Aragón C, López-Corcuera B. Calnexin-assisted biogenesis of the neuronal glycine transporter 2 (GlyT2). PLoS One 2013; 8: e63230.
↵ 108. Cubelos B, Giménez C, Zafra F. The glycine transporter GLYT1 interacts with Sec3, a component of the exocyst complex. Neuropharmacology 2005; 49: 935-44.
↵ 109. De Juan-Sanz J, Núñez E, Villarejo-López L, Pérez-Hernández D, Rodríguez-Fraticelli AE, López-Corcuera B, et al. Na+/K+-ATPase is a new interacting partner for the neuronal glycine transporter GlyT2 that downregulates its expression in vitro and in vivo. J Neurosci 2013; 28: 14269-81.
↵ 110. De Juan-Sanz J, Núñez E, Zafra F, Berrocal M, Corbacho I, Ibáñez I, et al. Presynaptic control of glycine transporter 2 (GlyT2) by physical and functional association with plasma membrane Ca2+-ATPase (PMCA) and Na+-Ca2+ exchanger (NCX). J Biol Chem 2014; 289: 34308-24.
↵ 111. Geerlings A, López-Corcuera B, Aragón C. Characterization of the interactions between the glycine transporters GLYT1 and GLYT2 and the SNARE protein syntaxin 1A. FEBS Lett 2000; 470: 51-4.
↵ 112. López-Colomé AM, Gadea A. Regulation of glycine transport in cultured Müller cells by Ca2+/calmodulin-dependent enzymes. Ann N Y Acad Sci 1999; 868: 685-8.
↵ 113. Núñez E, Alonso-Torres P, Fornés A, Aragón C, López-Corcuera B. The neuronal glycine transporter GLYT2 associates with membrane rafts: functional modulation by lipid environment. J Neurochem 2008; 105: 2080-90.
↵ 114. De Juan-Sanz J, Zafra F, López-Corcuera B, Aragón C. Endocytosis of the neuronal glycine transporter GLYT2: role of membrane rafts and protein kinase C-dependent ubiquitination. Traffic 2011; 12: 1850-67.
↵ 115. De Juan-Sanz J, Núñez E, López-Corcuera B, Aragón C. Constitutive endocytosis and turnover of the neuronal glycine transporter GlyT2 is dependent on ubiquitination of a C-terminal lysine cluster. PLoS One 2013; 8: e58863.
↵ 116. Mitchell K, Spike RC, Todd AJ. An immunocytochemical study of glycine receptor and GABA in laminae I-III of rat spinal dorsal horn. J Neurosci 1993; 13: 2371-81.
↵ 117. Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci 2009; 32: 1-32.
↵ 118. Kato G, Yasaka T, Katafuchi T, Furue H, Mizuno M, Iwamoto Y, et al. Direct GABAergic and glycinergic inhibition of the substantia gelatinosa from the rostral ventromedial medulla revealed by in vivo patch-clamp analysis in rats. J Neurosci 2006; 26: 1787-94.
↵ 119. Ossipov MH, Dussor GO, Porreca F. Central modulation of pain. J Clin Invest 2010; 120: 3779-87.
↵ 120. Von Hehn CA, Baron R, Woolf CJ. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron 2012; 73: 638-52.
↵ 121. Suhren O, Bruyn G, Tynman J. Hyperekplexia. A hereditary startle syndrome. J Neurol Sci 1966; 3: 577-605.
↵ 122. Sáenz-Lope E, Herranz-Tanarro FJ, Masdeu JC, Chacón-Peña JR. Hyperekplexia: a syndrome of pathological startle responses. Ann Neurol 1984; 15: 36-41.
↵ 123. Meinck HM. Startle and its disorders. Neurophysiol Clin 2006; 36: 357-64.
↵ 124. Rees MI, Harvey K, Pearce BR, Chung SK, Duguid IC, Thomas P, et al. Mutations in the gene encoding GlyT2 (SLC6A5) define a presynaptic component of human startle disease. Nat Genet 2006; 38: 801-6.
↵ 125. Eulenburg V, Retiounskaia M, Papadopoulos T, Gomeza J, Betz H. Glial glycine transporter 1 function is essential for early postnatal survival but dispensable in adult mice. Glia 2013; 58: 1066-73.
↵ 126. Giménez C, Pérez-Siles G, Martínez-Villarreal J, Arribas-González E, Jiménez E, Núñez E, et al. A novel dominant hyper-ekplexia mutation Y705C alters trafficking and biochemical properties of the presynaptic glycine transporter GlyT2. J Biol Chem 2012; 287: 28986-9002.
↵ 127. Applegarth DA, Toone JR. Glycine encephalopathy (nonketotic hyperglycinemia): comments and speculations. Am J Med Genet A 2006; 140: 186-8.
↵ 128. Mayor F Jr, Martín A, Rodríguez-Pombo P, García MJ, Benavides J, Ugarte M. Atypical nonketotic hyperglycinemia with a defective glycine transport system in nervous tissue. Neurochem Pathol 1984; 2: 233-49.
↵ 129. Jursky F, Nelson N. Developmental expression of the glycine transporters GLYT1 and GLYT2 in mouse brain. J Neurochem 1996; 67: 336-44.
↵ 130. Conter C, Rolland MO, Cheillan D, Bonnet V, Maire I, Froissart R. Genetic heterogeneity of the GLDC gene in 28 unrelated patients with glycine encephalopathy. J Inherit Metab Dis 2006; 29: 135-42.
↵ 131. Gether U, Andersen PH, Larsson OM, Schousboe A. Neuro-transmitter transporters: molecular function of important drug targets. Trends Pharmacol Sci 2006; 27: 375-83.
↵ 132. Sheldon AL, Robinson MB. The role of glutamate transporter in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int 2007; 51: 333-55.
↵ 133. Hermans H, Muth-Selbach U, Willians R, Kug S, Lipfert P, Werdehausen R, et al. Differential effects of spinally applied glycine transporters inhibitors on nociception in a rat model of neuropathic pain. Neurosci Lett 2008; 445: 214-9.
↵ 134. Carland JE, Handford CA, Ryan, RM, Vandenberg RJ. Lipid inhibitors of high affinity glycine transporters: identification of a novel class of analgesics. Neurochem Int 2014; 73: 211-6.
↵ 135. Dohi T, Morita K, Motoyama N, Morioka N. Glycine transporters inhibitors as a novel drug discovery strategy for neuropathic pain. Pharmacol Ther 2009; 123: 54-79.
↵ 136. Vandenberg RJ, Ryan RM, Carland JE, Imlach WL, Christie MJ. Glycine transport inhibitors for the treatment of pain. Trends Parmacol Sci 2014; 35: 423-30.
↵ 137. Armbruster A, Neumann E, Kötter V, Hermanns H, Werdehausen R, Eulenburg V. The GlyT1 inhibitor bitopertin ameliorates allodynia and hyperalgesia in animal models of neuropathic and inflammatory pain. Front Mol Neurosci 2018; 10: 438.
|
Pathophysiology of the glutamate and the glycine transporters: new therapeutic targets
Introduction. The amino acids glutamate and glycine, apart from their role in protein synthesis, are two fundamental neurotransmitters in the central nervous system of mammals. The first one is ubiquitous and is involved in excitatory pathways of the neocortex, the retina and the cerebellum, and the second is involved in inhibitory pathways of brain caudal areas. However, both share their way of acting by integrating into the functioning of glutamate receptors of the NMDA type fundamentals in the regulation of motor, sensory and cognitive systems.
Aim. To highlight the need for a fine regulation of glutamate and glycine concentrations in the intracellular and extracellular spaces of the nervous system through the action of very specific transporters for both neurotransmitters located in the plasma membrane of neurons and glial cells.
Development. The role of the glutamate and glycine transporters in glutamatergic and glycinergic neurotransmission and in the functioning of the nervous system is described. The pathological consequences of imbalances in these signaling pathways are pointed out. We also describe its involvement in pathologies such as schizophrenia, chronic pain, cerebral ischemia, diseases such as hereditary hyperekplexia and the non-ketotic hyperglycinemia, and neurodegenerative disorders.
Conclusions. The knowledge at molecular level of the way of acting of these transporters for glutamate and glycine is allowing the identification and development of new therapeutic strategies for pathologies such as those described above and the development of new drugs.
Key words. Cerebral ischemia. Chronic pain. Glutamate. Glycine. Hyperekplexia. Neurodegenerative disorders. Non-ketotic hyperglycinemia. Schizophrenia. Transporters. |
© 2018 Revista de Neurología
Organizado por:
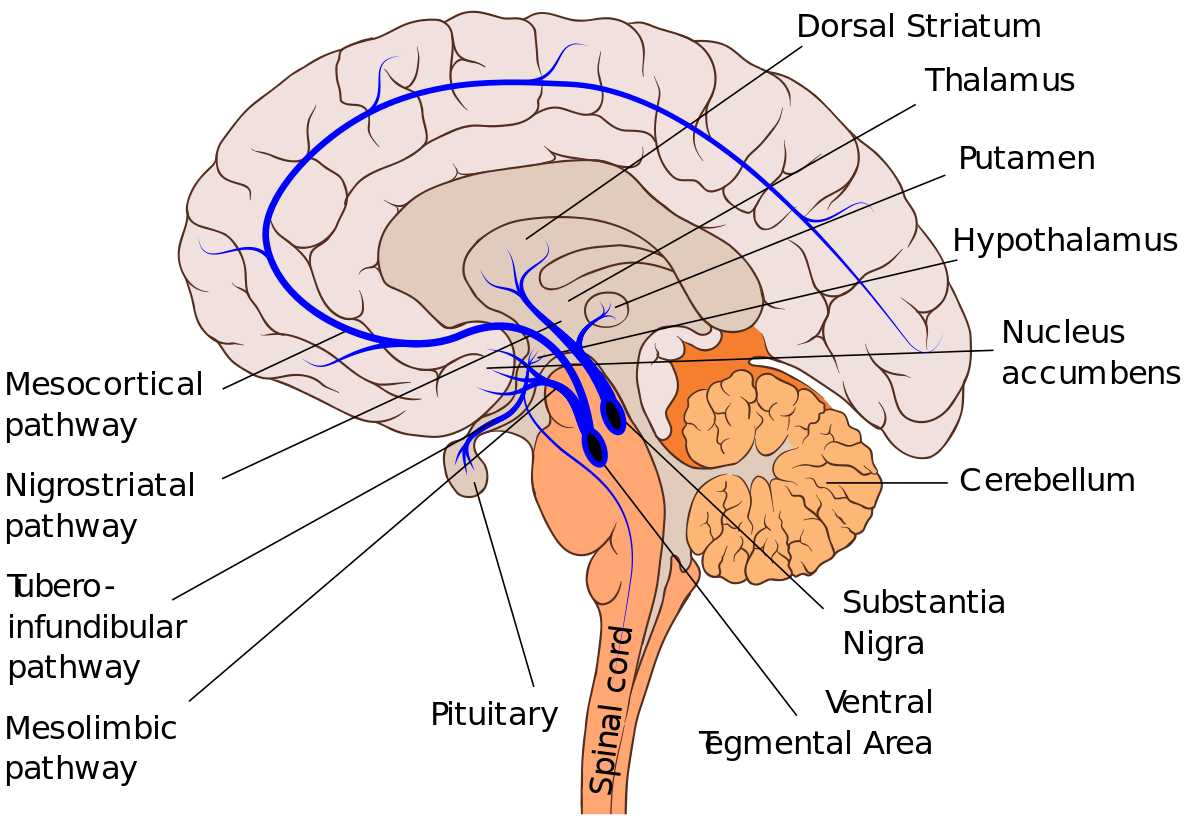
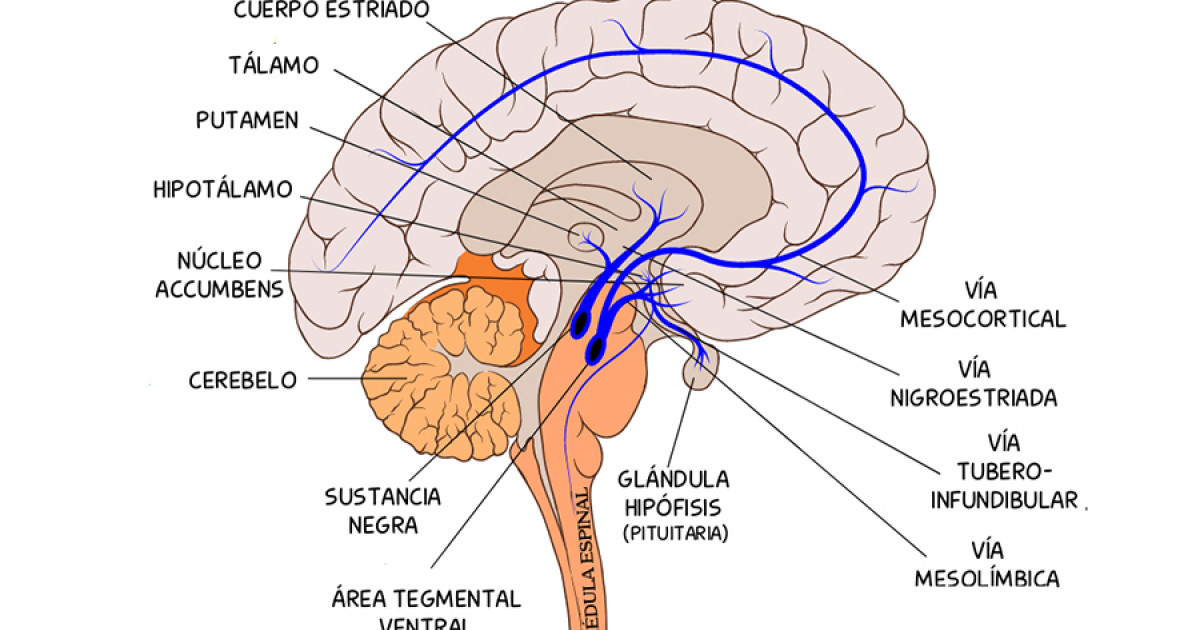 La vía nigroestriada es aquella cuyos haces se proyectan desde la sustancia negra al cuerpo estriado, concretamente al núcleo caudado y al putamen y tiene un papel fundamental en control motor, siendo la estimulación del movimiento intencional la principal función de esta.
La vía nigroestriada es aquella cuyos haces se proyectan desde la sustancia negra al cuerpo estriado, concretamente al núcleo caudado y al putamen y tiene un papel fundamental en control motor, siendo la estimulación del movimiento intencional la principal función de esta.