PLACAS DE AMILOIDE
26/05/2016 – 7:13
El Hospital General de Massachusetts (MGH,), evidencia que la proteína beta-amiloide (A-beta) se deposita en forma de placas en el cerebro de pacientes con enfermedad de Alzheimer– y se la considera una parte normal del sistema inmune innato, primera línea de defensa del cuerpo contra la infección.
Su estudio, publicado en ‘Science Translational Medicine’, concluye que la expresión de beta-amiloide humana resulta protectora contra las infecciones potencialmente letales en ratones, en gusanos redondos y en células cerebrales humanas en cultivo. «Se ha pensado que la neurodegeneración en la enfermedad de Alzheimer es causada por el comportamiento anormal de moléculas de beta-amiloide, que son conocidas por reunirse en resistentes estructuras de fibrillas llamadas placas amiloides en el cerebro de los pacientes», lo explica el coautor del artículo Robert Moir, de la Unidad de Genética y Envejecimiento del Instituto de Investigación de Enfermedades Neurodegenerativas del Hospital General de Massachusetts (MGH-MIND).
Un estudio de 2010 co-conducido por Moir y Rudolph Tanzi, director del MGH-MIND y co-autor correspondiente del trabajo actual, según la opinión de Moir , la beta-amiloide tenía muchas de las cualidades de un péptido antimicrobiano (AMP) y se trata de una pequeña proteína innata del sistema inmune que protege contra una amplia gama de patógenos.
En ese estudio se compararon formas sintéticas de A-beta con un conocido AMP llamado LL-37 y se encontró que A-beta inhibe el crecimiento de varios patógenos importantes, a veces igual de bien o mejor que LL-37. A-beta de los cerebros de los pacientes de Alzheimer también suprimió el crecimiento del hongo ‘Candida’ cultivado para esa investigación y, posteriormente, otros grupos han documentado la acción de A-beta sintético contra los virus de influenza y herpes.
En este nuevo trabajo, los investigadores encontraron que los ratones transgénicos que expresan A-beta humano sobrevivieron significativamente más tiempo después de inducir la infección por ‘Salmonella’ en sus cerebros frente a los ratones sin alteración genética. Los ratones que carecen de la proteína precursora de amiloide murieron incluso más rápidamente.
La expresión de A-beta transgénica también parece proteger a los gusanos redondos ‘C.elegans’ de cualquier infección por ‘Candida’ o ‘Salmonella’. Del mismo modo, la expresión de A-beta humana protegión las células neuronales cultivadas de ‘Candida’. De hecho, el A-beta humano expresado por células vivas parece ser mil veces más potente contra la infección que el A-beta sintético utilizado en estudios previos.
Esa superioridad parece referirse a propiedades de A-beta que se han considerado parte de la patología en la enfermedad de Alzheimer, la propensión de moléculas pequeñas a combinarse en lo que se denominan oligómeros y luego se agregan en placas de beta-amiloide.
Mientras los AMP combaten la infección a través de varios mecanismos, un proceso fundamental implica la formación de oligómeros que se unen a las superficies microbianas y luego se agrupan en agregados que evitan que los patógenos se adhieran a las células huésped y permiten a los AMP matar microbios mediante la interrupción de sus membranas celulares.
Las preparaciones sintéticas de A-beta utilizadas en los estudios anteriores no incluyeron oligómeros; pero en el actual estudio, el oligómero de A-beta humano no sólo mostró una actividad antimicrobiana aún más fuerte, sino que también se observó su agregación en los tipos de fibrillas que forman las placas de beta-amiloide para atrapar los microbios en los modelos de ratones y gusanos redondos.
Tanzi explica que «se sabe que AMP juega un papel en las patologías de una amplia gama de enfermedades inflamatorias». «Por ejemplo, LL-37, que ha sido nuestro modelo para la actividad antimicrobiana de A-beta, ha sido implicado en varias enfermedades al final de la vida, incluyendo artritis reumatoide, lupus y aterosclerosis. El tipo de desregulación de la actividad de AMP que puede causar inflamación mantenida en esos trastornos podría contribuir a las acciones neurodegenerativas de A-beta en la enfermedad de Alzheimer» señala.
Moir añade: «Nuestros resultados plantean la intrigante posibilidad de que puede surgir la patología de Alzheimer cuando el cerebro se percibe a sí mismo como bajo el ataque de los patógenos invasores, aunque se necesitan estudios adicionales. No parece probable que las vías inflamatorias del sistema inmune innato puedan ser posibles dianas de tratamiento. Si se validan, nuestros datos también justifican la necesidad de tener precaución con terapias dirigidas a la eliminación total de las placas de beta-amiloide. Las terapias basadas en la disminución de amiloides, pero no en la eliminación de beta amiloide en el cerebro podría ser una estrategia mejor».
El siguiente paso es la búsqueda de microbios en los cerebros de pacientes de Alzheimer que puedan haber desencadenado la deposición de amiloide como una respuesta protectora, llevando más tarde a la muerte de las células nerviosas y la demencia. «Si podemos identificar a los culpables –ya sean bacterias, virus u hongos– podemos ser capaces de dirigirnos a ellos terapéuticamente para la prevención primaria de la enfermedad», concluye Tanzi
Categoría: SINUCLEOPATIAS
 La enfermedad de Parkinson y la inmunidad :
La enfermedad de Parkinson y la inmunidad :
Fue descrita por primera vez en 1817 por James Parkinson en su Ensayo sobre la Parálisis Temblorosa. SJames Parkinson in his Essay on the Shaking Palsy.
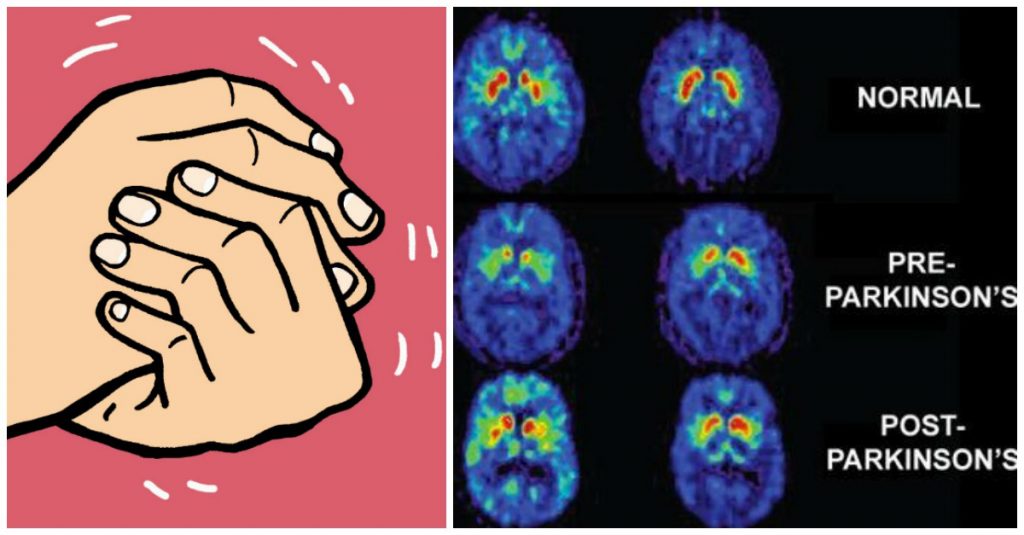
La enfermedad de Parkinson es una enfermedad crónica caracterizada principalmente por una pérdida progresiva de la capacidad de coordinar los movimientos. Esto se debe a la degeneración y muerte progresiva de unas células nerviosas (neuronas) situadas principalmente en una pequeña parte del cerebro conocida como sustancia negra. Dichas neuronas se encargan de producir la dopamina, una sustancia que permite que las células cerebrales implicadas en el control del movimiento se puedan comunicar entre sí. Cuando los niveles de dopamina bajan, dicha comunicación no se desarrolla correctamente, provocando patrones anormales de activación nerviosa lo que se traduce en temblor, rigidez, lentitud de movimiento y trastornos posturales, síntomas comunes en la enfermedad de Parkinson. Otra de las características de la enfermedad de Parkinson es la acumulación de formas defectuosas de la proteína alfa-sinucleína y la formación de los así llamados cuerpos de Lewy en las neuronas productoras de dopamina.
Cada vez hay más indicios de que reacciones desreguladas del sistema inmune y la inflamación crónica en el sistema nervioso central pueden contribuir al avance de esta enfermedad.
Aunque tradicionalmente el sistema nervioso central (SNC) se consideró como un tejido “inmunoprivilegiado”, en el que el sistema inmune periférico no podía acceder, evidencias clínicas y experimentales han demostrado la relación interactiva entre estos dos sistemas. El sistema inmune es responsable de la protección del SNC ante infecciones o el daño cerebral. Cuando las células microgliales, que representan las células del sistema inmune innato más importantes en el cerebro, detectan agentes infecciosos y restos de células dañadas, se activan y liberan citóquinas proinflamatorias y otras sustancias que inducen un proceso inflamatorio, aumentando la permeabilidad de la barrera entre los vasos sanguíneos y el sistema nervioso central (barrera hematoencefálica) y permitiendo la entrada de linfocitos T en el SNC. Hay evidencias de que no sólo agentes infecciosos y restos celulares pueden inducir una respuesta por parte del sistema inmune innato y adaptativo, sino también las proteínas modificadas en el cerebro como la alfa-sinucleína que mencionamos al principio.
En condiciones normales, esta reacción es esencial para eliminar el tejido dañado y restablecer el equilibrio y las funciones normales del SNC. Sin embargo, una inflamación persistente y una activación incontrolada de células del sistema inmune puede ser perjudicial para el SNC y puede iniciar o amplificar la neurodegeneración. Es por ello que es importante regular estos procesos en la enfermedad de Parkinson.
Parkinson familiar y demencia con cuerpos de Lewy: un estudio de secuenciación y vinculación de genoma completo
•
La mayoría de los pacientes con enfermedad de Parkinson, demencia por enfermedad de Parkinson y demencia con cuerpos de Lewy no portan mutaciones en genes conocidos que causan enfermedades. El objetivo de este estudio fue identificar un nuevo gen implicado en el desarrollo de estos trastornos.
Nuestro estudio se realizó en tres etapas. Primero, hicimos un análisis de ligamiento genómico de una familia italiana con enfermedad de Parkinson dominantemente heredada para identificar el locus de la enfermedad. En segundo lugar, secuenciamos el gen candidato en una serie internacional multicéntrica de probandos no emparentados que fueron diagnosticados clínica o patológicamente con la enfermedad de Parkinson, la demencia de la enfermedad de Parkinson o la demencia con cuerpos de Lewy. Como control, utilizamos datos de secuenciación de genes de individuos con aneurismas aórticos abdominales (que no fueron examinados neurológicamente). En tercer lugar, inscribimos una serie independiente de pacientes diagnosticados clínicamente con enfermedad de Parkinson y controles sin signos o antecedentes familiares de enfermedad de Parkinson, demencia por enfermedad de Parkinson o demencia con cuerpos de Lewy de centros en Portugal, Cerdeña y Taiwán, y los seleccionó para variantes específicas. También realizamos estudios de mRNA y patología cerebral en tres pacientes de la serie internacional multicéntrica portadora de variantes asociadas a la enfermedad, e hicimos estudios de proteínas funcionales en modelos in vitro, incluidas las neuronas de células pluripotentes inducidas de tallo.
Los estudios moleculares se realizaron entre el 1 de enero de 2008 y el 31 de diciembre de 2017. En el parentesco inicial de diez individuos italianos afectados (edad media de aparición de la enfermedad 59 · 8 años [SD 8 · 7]), detectamos un vínculo significativo de la enfermedad de Parkinson al cromosoma 14 y nominado LRP10 como el gen causante de la enfermedad. Entre las series internacionales de 660 probandos, identificamos ocho individuos (cuatro con enfermedad de Parkinson, dos con demencia por enfermedad de Parkinson y dos con demencia con cuerpos de Lewy) que portaban LRP10 diferente, raro y potencialmente patogénico .variantes; un portador se encontró entre 645 controles con aneurismas aórticos abdominales. En la serie independiente, se detectaron dos de estas ocho variantes en tres probandos adicionales de la enfermedad de Parkinson (dos de Cerdeña y uno de Taiwán) pero en ninguno de los controles. De los 11 probandos de las cohortes internacionales e independientes con variantes de LRP10 , diez tenían un historial familiar positivo de la enfermedad y el ADN estaba disponible de diez familiares afectados (en siete de estas familias). Las variantes de LRP10 estuvieron presentes en nueve de estos diez parientes, proporcionando evidencia independiente, aunque limitada, de co-segregación con la enfermedad. Estudios post-mortem en tres pacientes con LRP10 distintovariantes mostraron patología corporal de Lewy severa. De nueve variantes identificadas en total (una en la familia inicial y ocho en la etapa 2), tres severamente afectadas expresión de LRP10 y estabilidad del mRNA (1424 + 5delG, 1424 + 5G → A, y Ala212Serfs * 17, mostradas por análisis de cDNA), cuatro la estabilidad de la proteína afectada (Tyr307Asn, Gly603Arg, Arg235Cys y Pro699Ser, mostrada por cycloheximide-chase experimentos), y dos localización de la proteína afectada (Asn517del y Arg533Leu, demostrado por inmunocitoquímica), apuntando a la pérdida de la función LRP10 como mecanismo patogénico común.
La Interpretación de nuestros hallazgos implican defectos del gen LRP10 en el desarrollo de formas heredadas de α-sinucleinopatías. La elucidación futura de la función de la proteína LRP10 y las vías podría ofrecer nuevos conocimientos sobre los mecanismos, biomarcadores y objetivos terapéuticos.
En Stichting ParkinsonFonds, Dorpmans-Wigmans Stichting, Erasmus Medical Center, programa ZonMw-Memorabel, programa conjunto de la UE Neurodegenerative Disease Research (JPND), Parkinson’s UK, Avtal om Läkarutbildning och Forskning (ALF) y Parkinsonfonden (Suecia), Lijf y Leven foundation, y concesión transfronteriza de Alzheimer Netherlands-Ligue Européene Contre la Maladie d’Alzheimer (LECMA).
REGULACIÓN DE LOS CANALES DE CALCIO
Redacción. Madrid | 2018-06-13 12:42:59
Eliminar los desechos de proteínas que afectan a las neuronas, potencial clave para Parkinson
Científicos de la UGR y el CSIC han llegado a la conclusión de que nuevos métodos que favorezcan la eliminación de los agregados de proteínas que los enfermos de Parkinson acumulan podrían constituir una estrategia terapéutica prometedora contra esta patología.
La investigadora de la UGR Pilar Rivero Ríos, en el centro de la imagen, junto a su equipo.
Conseguir fármacos que ataquen la causa del Parkinson y no se limiten solo a aliviar sus síntomas es el objetivo final de la investigación de Pilar Rivero Ríos, doctoranda de la Universidad de Granada que realiza su tesis en el grupo de Sabine Hilfiker, en el Instituto López-Neyra del CSIC, en Granada. Para ello, los científicos han llegado a la conclusión de que nuevas aproximaciones para promover la eliminación de los agregados de proteínas que los enfermos de Parkinson acumulan podrían constituir una estrategia terapéutica prometedora contra esta patología, según publican en Messenger.
El Parkinson es la segunda enfermedad neurodegenerativa más común, ya que afecta al 1 por ciento de la población mayor de 65 años. Uno de sus rasgos característicos es la presencia de agregados de proteínas que, en circunstancias normales, deberían ser desechados, y que se acumulan hasta provocar la muerte de las neuronas. «Este hecho apunta a la existencia de alteraciones en los lisosomas, que podrían compararse con el ‘aparato digestivo’ de la célula»,
LRRK2 en el lisosoma
Los científicos analizan los mecanismos mediante los cuales LRRK2, principal determinante genético del Parkinson, provoca dicha enfermedad al afectar a los canales de calcio que se encuentran en el lisosoma, impidiendo que este realice su función de eliminación de los desechos que finalmente provocan la muerte celular. Conocer los mecanismos responsables de la enfermedad es el primer paso para el desarrollo de fármacos eficaces para tratarla.
«Los tratamientos frente al Parkinson de los que disponemos en la actualidad presentan el problema de que se limitan a aliviar los síntomas, pero no atacan a la causa y por tanto no curan. De ahí la importancia de conocer cuáles son los procesos que están afectados en la célula, lo que permitirá desarrollar fármacos que corrijan esos procesos y realmente curen la enfermedad en lugar de simplemente atacar los síntomas. La investigación sería un primer paso para el desarrollo de fármacos frente al Parkinson», Rivero.
La acumulación en las neuronas de desechos celulares, los llamados cuerpos de Lewy, es uno de los rasgos característicos de la enfermedad de Parkinson. La investigadora explica que «el desarrollo de fármacos que regulen la actividad de estos canales de calcio que se encuentran alterados en el Parkinson podría corregir el mal funcionamiento del lisosoma y combatir la patología». En definitiva, se atacarían las causas del Parkinson y no simplemente sus síntomas.
Sin embargo, alerta de la complejidad del proceso de creación de nuevos fármacos: «Solo un 1 por ciento de los fármacos de nueva creación superan los controles de eficacia, seguridad en laboratorio y los ensayos clínicos. Puede pasar una década desde el desarrollo del fármaco hasta que está disponible para las personas que lo necesitan».
Resumen
lLa enfermedad de Parkinson es crónica y se caracterizada principalmente por una pérdida progresiva de la capacidad de coordinar los movimientos. Esto se debe a la degeneración y muerte progresiva de las neuronas situadas principalmente en una pequeña parte del cerebro conocida como sustancia negra. Dichas neuronas se encargan de producir la dopamina, una sustancia que permite que las células cerebrales implicadas en el control del movimiento se puedan comunicar entre sí. Cuando los niveles de dopamina bajan, dicha comunicación no se desarrolla correctamente, provocando patrones anormales de activación nerviosa lo que se traduce en temblor, rigidez, lentitud de movimiento y trastornos posturales, síntomas comunes en la enfermedad de Parkinson.
Otra de las características de la enfermedad de Parkinson es la acumulación de formas defectuosas de la proteína alfa-sinucleína y la formación de los así llamados cuerpos de Lewy en las neuronas productoras de dopamina
La acumulación en las neuronas de desechos celulares, los llamados cuerpos de Lewy, es uno de los rasgos característicos de la enfermedad de Parkinson. Un eje de un
La mayoría de los pacientes con enfermedad de Parkinson, demencia por enfermedad de Parkinson y demencia con cuerpos de Lewy no portan mutaciones en genes conocidos que causan enfermedades.
La YLRRK2, principal determinante genético del Parkinson, provoca dicha enfermedad al afectar a los canales de calcio que se encuentran en el lisosoma, impidiendo que este realice su función de eliminación de los desechos que finalmente provocan la muerte celular
Referencia:
R. Lee Mosley, Jessica A. Hutter-Saunders, David K. Stone, and Howard E. Gendelman. Inflammation and Adaptive Immunity in Parkinson’s Disease. Cold Spring Harb Perspect Med. Jan 2012; 2(1): a009381.
REGULACIÓN DE LOS CANALES DE CALCIO
Redacción. Madrid | 2018-06-13 12:42:59
Eliminar los desechos de proteínas que afectan a las neuronas, potencial clave para Parkinson
Parkinson familiar y demencia con cuerpos de Lewy: un estudio de secuenciación y vinculación de genoma completo
SINUCLEOPATIAS
El término a-sinucleinopatía se utiliza para referirse a un grupo de enfermedades que tienen en común el depósito anormal de a-sinucleína en el citoplasma de neuronas o de células gliales, o en el neurópilo. En la enfermedad de Parkinson y en la demencia de cuerpo de Lewy, los depósitos de a-sinucleína constituyen el componente principal de los cuerpos de Lewy y de las neuritas distróficas; también, en menor proporción, la a-sinucleína se deposita en el citoplasma de células gliales. En la atrofia multisistémica, la a-sinucleína se acumula en las inclusiones citoplásmicas de las células oligodendrogliales y en las neuronas, así como en las neuritas distróficas en el tronco del encéfalo. Finalmente, el fragmento medio de 61-95 aminoácidos de a-sinucleína es el componente no-Aß del amiloide de las placas seniles en la enfermedad de Alzheimer. Los depósitos de a-sinucleína en estas enfermedades tienen en común la configuración fibrilar, pero difieren en la unión de a-sinucleína con distintas proteínas en cada una de las enfermedades, con la excepción de las ubicuitinas que se encuentran en las distintas inclusiones. No se conoce el mecanismo por el que la a-sinucleína se fragmenta, se libera al espacio extracelular y forma el 10% del amiloide de las placas seniles. Interacciones de ßA4 y a-sinucleína pueden explicar la unión de estos productos resultantes de la fragmentación anómala de proteínas precursoras. Por otra parte, distintos estudios han demostrado que la a-sinucleína puede formar estructuras fibrilares y dar lugar a formas insolubles y a agregados de alto peso molecular in vitro y en las a-sinucleinopatías. Aunque estudios in vitro han señalado el efecto tóxico de las fibrillas de a-sinucleína, no se conoce el efecto del depósito de la a-sinucleína sobre la supervivencia celular en las a-sinucleinopatías.
Neurología 2001;16(4): 163-170 Ferrer