Varias veces en la literatura se ha relacionado con gérmenes y macrófagos al enfermedad de AlZHEIMER
El descubrimiento del patógeno que provoca la periodontitis crónica en el cerebro de pacientes fallecidos de Alzheimer puede revolucionar los estudios sobre esta enfermedad Un nuevo estudio sugiere que una mala salud bucal podría provocar Alzheimer
A lo largo de los últimos años, diversos científicos han apuntado a una hipótesis poco conocida sobre el origen del Alzheimer: según su teoría no sería sólo una enfermedad, sino una infección. Ahora, un nuevo estudio publicado en Science Advances refuerza esa teoría y lo hace vinculando esa enfermedad degenerativa con un origen inesperado: una enfermedad en las encías.
Jan Potempa, microbiólogo de la Universidad de Louisville, ha descubierto ‘Porphyromonas gingivalis’ en el cerebro de pacientes fallecidos de Alzheimer. Se trata del patógeno que provoca la periodontitis crónica, conocida como enfermedad de las encías, y no es la primera vez que ambas enfermedades se asocian, ya que un estudio de 2010 ya valoró esa posibilidad.
Se han llevado a cabo experimentos con ratones, en los que se provocaban infecciones orales con el patógeno, que condujeron a que las bacterias llegaran al cerebro y que éste produjera el péptido beta amiloide (Aβ), la proteína asociada con el Alzheimer. Los científicos creen que aún no han descubierto el origen de esta enfermedad degenerativa, pero están convencidos de que esta línea de investigación es importante. A. López
Stephen Dominy, que coordina la investigación a través de la farmacéutica Cortexyme, asegura a Science Alert que «los agentes infecciosos han estado implicados en el desarrollo y la progresión del Alzheimer antes, pero la evidencia de la causalidad no había sido convincente. Ahora, por primera vez, tenemos pruebas sólidas que conectan el patógeno intracelular P. gingivalis y la enfermedad de Alzheimer».
La importancia de la boca
Los investigares no se quedaron ahí e identificaron enzimas tóxicas llamadas gingipainas en el cerebro de pacientes con Alzheimer. Pero, además, también encontraron esas gingipainas en cerebros de personas fallecidas que no fueron diagnosticadas con Alzheimer.
No es la primera vez que se relaciona la infección la inflamación y el deposito de proteínas en el cerebro
El Alzheimer (AL) es una enfermedad neurodegenerativa, y la causa principal y más conocida en enfermedades demenciales, cuya característica principal es la pérdida de memoria. Su relación con la proteína amiloide y tau es indiscutible. Su acumulo en el lóbulo temporal entre otras zonas del cerebro mutila la funciones cognoscitiva y produce demencia. La Esclerosis Múltiple, (EM) también es una enfermedad neurodegenerativa, y afecta a los nervios, destruye la mielina que es su aislante Los nervios están envueltos en mielina que los aísla y permite su función. La beta-amiloide sería capaz de luchar contra la reacción inflamatoria autoinmune que causa la EM, la proteína beta-amiloide y su precursor se encuentran en las lesiones de la EM .
La administración de proteínas beta-amiloide fuera del cerebro, concretamente se inyectó en el vientre de los ratones y no en el cerebro. Aunque los sistemas inmunes de estos ratones estaban preparados para atacar al aislante nervioso o mielina, con la administración de la beta-amiloide se produjo todo lo contrario.
Así lo expresa Steinman: “Esta es la primera vez que la beta amiloide demuestra tener propiedades antiinflamatorias” En definitiva, la beta-amiloide fuera del sistema nervioso central tendría propiedades como antiinflamatorio, todo lo contrario a lo que produce cuando se encuentra dentro del mismo cerebro o de los nervios. El juego inflamatorio en enfermedades degenerativas, llegan siempre a las misma situaciones, un problema de inflamación y reparación complejo e imbricado. No es la primera vez que se analizan las propiedades de la beta amiloide, El Hospital General de Massachusetts (MGH,), evidencio que la proteína beta-amiloide se deposita en forma de placas en el cerebro de pacientes con enfermedad de AL y se la considera una parte normal del sistema inmune innato, primera línea de defensa del cuerpo contra la infección. Su estudio, publicado en ‘Science Translational Medicine’, concluye que la expresión de beta-amiloide humana resulta protectora contra las infecciones potencialmente letales en ratones, en el C elegans y en células cerebrales humanas en cultivo.
Se admite que la neurodegeneración en la enfermedad de AL es causada por el comportamiento anormal de moléculas de beta-amiloide, que son conocidas por reunirse en resistentes estructuras de fibrillas llamadas placas amiloides en el cerebro de los pacientes En 2010 , Moir y Rudolph Tanzi, director del MGH-MIND y concluyeron que la beta-amiloide tenía muchas de las cualidades de un péptido antimicrobiano (AMP) y se trata de una pequeña proteína innata del sistema inmune que protege contra una amplia gama de patógenos. En ese estudio se compararon formas sintéticas de A-beta con un conocido AMP llamado LL-37 y se encontró que la beta inhibe el crecimiento de varios patógenos importantes, a veces igual de bien o mejor que LL-37. La beta amiloide de los cerebros de los pacientes de AL también suprimió el crecimiento del hongo ‘Candida’ cultivado para esa investigación y, posteriormente, otros grupos han documentado la acción de A-beta sintético contra los virus de influenza y herpes. En este nuevo trabajo, los investigadores encontraron que los ratones transgénicos que expresan A-beta humano sobrevivieron significativamente más tiempo después de inducir la infección por ‘Salmonella’ en sus cerebros frente a los ratones sin alteración genética. Los ratones que carecen de la proteína precursora de amiloide murieron incluso más rápidamente. La expresión de la beta transgénica parece proteger a los gusanos ‘C.elegans’ de cualquier infección por ‘Candida’ o ‘Salmonella’. Del mismo modo, la expresión de beta humana protege las células neuronales cultivadas de ‘Candida’. De hecho, la beta amiloide humano expresado por células vivas parece ser mil veces más potente contra la infección que el beta amiloide sintético utilizado en estudios previos. Esa superioridad parece referirse a propiedades de beta amiloides que se han considerado parte de la patología en la enfermedad AL, la propensión de moléculas pequeñas a combinarse en lo que se denominan oligómeros y luego se agregan en placas de beta-amiloide.
Moir añade: “Nuestros resultados plantean la intrigante posibilidad de que puede surgir la patología de AL, cuando el cerebro se percibe a sí mismo como bajo el ataque de los patógenos invasores, aunque se necesitan estudios adicionales. No parece probable que las vías inflamatorias del sistema inmune innato puedan ser posibles dianas de tratamiento. Si se validan, nuestros datos también justifican la necesidad de tener precaución con terapias dirigidas a la eliminación total de las placas de beta-amiloide. Las terapias basadas en la disminución de amiloides, pero no en la eliminación de beta amiloide en el cerebro podría ser una estrategia mejor”.
Un nuevo estudio de la Universidad de Florida, en Estados Unidos, establece, por primera vez, un vínculo entre especies específicas de bacterias y manifestaciones físicas de enfermedades neurodegenerativas.
Investigaciones recientes sugieren que las personas con estas enfermedades presentan cambios en la composición bacteriana de su tracto digestivo.
Una nueva investigación publicada en ‘PLOS Pathogens’ establece, por primera vez, un vínculo entre especies específicas de bacterias y manifestaciones físicas de enfermedades neurodegenerativas.
En este estudio, quiere demostrar que especies específicas de bacterias desempeñan un papel en el desarrollo de estas enfermedades», Daniel Czyz,
Algunas otras bacterias producen compuestos que contrarrestan estas bacterias ‘malas’. Estudios recientes han demostrado que los pacientes con la enfermedad de Parkinson y de Alzheimer son deficientes en estas bacterias ‘buenas’, por lo que nuestros hallazgos pueden ayudar a explicar esa conexión y abrir un área de estudio futuro», añade.
Lo que se deduce de estos estudios, es que el deposito de proteínas mal plegadas, se hace sobre gérmenes previos y no al revés.
Varios autores sobre todo Carrasco y Moir, han repetido de forma machacona, como los gérmenes están presentes en las enfermedades neurodegenerativas y que los depositos de proteínas plegadas que son macrófagos es un intento del
Gérmenes en el Alzheimer
BIBLIOGRAFÍA
Sociedad Española de Medicina de Familia y Comunitaria. Demencias desde la Atención Primaria. Barcelona: semFYC; 2005. 2. Grupo de trabajo de la Guía de Práctica Clínica sobre la atención integral a las personas con enfermedad de Alzheimer y otras demencias. Guía de Práctica Clínica sobre la atención integral a las personas con enfermedad de Alzheimer y otras demencias. Plan de Calidad para el Sistema Nacional de Salud del Ministerio de Sanidad, Política Social e Igualdad. Agència d´Informació, Avaluació i Qualitat en Salut de Cataluña; 2010. Guías de Práctica Clínica en el SNS: AIAQS Núm. 2009/07. http://portal.guiasalud.es/web/guest/home 3. Hort J, O’Brien JT, Gainotti G, Pirttila T, Popescu BO, Rektorova I, Sorbi Sand and Scheltensh P. EFNS guidelines for the diagnosis and management of Alzheimer´s disease. Eur J Neurol 2010; 17: 1236-1248. 4. National Institute for Health and Clinical Excellence (NICE). Clinical guideline 42. Dementia: supporting people with dementia and their carers in health and social care. November 2006 (last modified: March 2011). http://www.nice.org.uk/CG42 5. Feldman HH, Jacova C, Robillard A, et al. Diagnosis and treatment of dementia: 2. Diagnosis. CMAJ 2008;178: 825-36. http://www.ecmaj.ca/cgi/content/abstract/178/7/825 6. Knopman DS, DeKosky ST, Cummings JL, et al. Practice parameter: diagnosis of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001: 56:1143-1153. http://www.neurology.org/content/56/9/1143.full.pdf+html 7. Olazaran J, Alteración cognitiva leve en la práctica clínica. Med Clin. 2011; 414-418. 8. Albert MS et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7: 270-9. 9. Robles A, Del Ser T, Alom J, Peña-Casanova J y Grupo Asesor del Grupo de Neurología de la Conducta y Demencias de la Sociedad Española de Neurología. Propuesta de criterios para el diagnóstico clínico del deterioro cognitivo ligero, la demencia y la enfermedad de Alzheimer. Neurología. 2002; 17(2): 17-32. 2 10. Chertkow H, Massoud F, Nasreddine Z, et al. Diagnosis and treatment of dementia: 3. Mild cognitive impairment and cognitive impairment without dementia. CMAJ. 2008;178:1273-85. http://www.cmaj.ca/cgi/content/abstract/178/10/1273 11. Petersen RC, Stevens JC, Ganguli M et al. Practice parameter: Early detection of dementia: Mild cognitive impairment (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001; 56: 1133-1142. http://www.neurology.org/content/56/9/1133.full.pdf+html 12. Petersen RC, Doody R, Kurz A, et al. Current concepts in mild cognitive impairment. Arch Neurol. 2001; 58:1985–1992 13. Contador I, Fernández-Calvo B, Ramos F, Tapias-Merino E, Bermejo-Pareja F. El cribado de la demencia en atención primaria. Revisión crítica. Rev Neurol. 2010; 51: 677-86. http://www.revneurol.com/sec/resumen.php?id=2010453# 14. Villarejo A, Puertas-Martín V. Utilidad de los test breves en el cribado de demencia. Neurología. 2011; 26 (7): 425-433 15. De Hoyos Alonso, Tapias Merino E, García de Blas F. Demencia. En: Los principales problemas de salud. AMF 2012; 8(9): 484-495. 16. Olazarán. J. Bermejo F: ¿Puede diagnosticarse la demencia en Atención Primaria? Aten Primaria. 2011; 43 (7): 377-384 17. Simmons B, Hartmann B, Dejoseph D. Evaluation of suspected dementia.. Am Fam Physician. 2011 Oct 15;84(8):895-902. 18. Riu S y Martínez A. Síndrome confusional agudo en el mayor. AMF. 2008; 4(4):216-21 19. American Geriatrics Society Updated Beers criteria for potentially Inappropriate medications use in older adults. American Geriatrics Society 2012 Beers Criteria Update Expert panel. JAGS 2012 http://www.americangeriatrics.org/files/documents/beers/2012BeersCriteria_JAGS.pdf 20. Riu S, Martínez A y Baena D. Fármacos que pueden alterar el estado cognitivo en el anciano. FMC.2009;16(5):
Alzheimer y Robert Moir « Enriquerubio.net
Luis Carrasco,. Catedrático de Microbiología de la UAM
Hasta ahora, el patógeno Porphyromonas gingivalis y el Alzheimer se habían relacionado pero no se sabía si la enfermedad de las encías provoca Alzheimer o si es la demencia la que conduce a un cuidado bucal deficiente. Ahora, el hecho de encontrar gingipainas en personas que nunca fueron diagnosticadas de Alzheimer podría sugerir que podrían haber desarrollado la enfermedad si hubieran vivido más tiempo.
O. Rodríguez 24/01/2019 – 12:47 Actualizado: 24/01/2020 – 15:24
Los autores de la investigación aseguran en su estudio que «nuestra identificación de antígenos de gingipaina en los cerebros de personas con enfermedad de Alzheimer y también con patología de esta enfermedad pero sin haber sido diagnosticados de demencia, sostiene que la infección cerebral con Porphyromonas gingivalis no es el resultado de una atención dental deficiente después del inicio de la demencia o una consecuencia de la enfermedad, sino un evento temprano que puede explicar la patología encontrada en personas de mediana edad antes del deterioro cognitivo».
Es el primer tratamiento contra esa enfermedad neurodegenerativa en casi 20 años
Según la Agencia del Medicamento, Aduhelm supone «un significativo avance»
La noticia ha sido recibida con cierto recelo por parte de un comité asesor independiente y algunos especialistas porque consideran que no hay aún suficiente evidencia de que el fármaco ayude a los pacientes
Estados Unidos ha aprobado un nuevo medicamente para tratar el Alzhéimer que afecta a la memoria y causa desorientación e impide, a veces, realizar tareas cotidianas. También se asocia con marcados cambios de humor y problemas de comunicación.
Es el primer tratamiento contra esa enfermedad neurodegenerativa en casi 20 años. En todo el mundo la padecen alrededor de cincuenta millones de personas y generalmente comienza después de los 65 años.
El fármaco se llama ‘Adulhem’ y según la Agencia del Medicamento supone un «gran avance». Es la primera medicina creada para tratar directamente la patología fundamental de este mal neurodegenerativo. Es decir, el fármaco reduce las placas beta en el Alzheimeramiloide en el cerebro, que son el signo más palpable de la enfermedad por culpa de su toxicidad.
Una teoría científica asegura que la enfermedad se produce por una acumulación excesiva de estas proteínas en el cerebro de algunas personas a medida que envejecen y su sistema inmunológico se deteriora.
Es el primero en abordar el deterioro cognitivo relacionado con la afección.
afectando la memoria de las personas, dejándolas desorientadas y en ocasiones incapaces de realizar las tareas cotidianas. También se asocia con marcados cambios de humor y problemas de comunicación.
La noticia ha sido recibida con cierto recelo por parte de un comité asesor independiente y algunos especialistas en esta enfermedad. Para ellos, aún no hay suficiente evidencia de que el fármaco ayude a los pacientes. La Agencia del Medicamento reconoce que los resultados no son concluyentes todavía pero considera que hay evidencia sustancial para su aprobación.
Este avance coincide con el avanzado estudio de investigadores del Alzheimer Center de Barcelona que han identificado nuevas variantes genéticas implicadas en la demencia senil de tipo Alzhéimer. Lo que hace el nuevo medicamento es actuar como un anticuerpo monoclonal, que elimina a la proteína macrofagica, Beta amiloidotica
Y eso parace eficaz en algunos casos.
Ya es un logro, pero estamos matando al anticuerpo que en el Alzheimer luchan contra algo. Hace falta seguir poniendo la atención en los gérmenes que desencadenan la acción de los macrófagos.
Pero si reconocemos que destruyendo los acúmulos de esta proteína, los pacientes mejoran. Habrá que pensar si esta medicación tan cara es soportable.
La otra cosa es buscar si de verdad los gérmenes son los responsables de que se deposite esta beta amiloide.
No me canso de decir que estas enfermedades neuro degenerativas están producidas por uno o varios gérmenes como vienen diciendo desde hace mucho tiempo los Dres. Carrasco y Moir
Gérmenes en el Alzheimer
Células de levaduras en depósitos de Amiloide
Gérmenes en el cerebro procedentes del intestino
Es lo lógico, los macrófagos, que actúan como anticuerpos se precipitan siempre que pueden sobre los gérmenes.
Si pudiéramos ver que razonablemente desaparecen la placas beta amiloidotica y reaparecen las condiciones psíquicas. Pues que bien.
Pero sobre todo correlacionar, enfermedades degenerativas con gérmenes y macrófagos
Pero además insisto en la necesidad de seguir pensando en los gérmenes como.
Lo que llama profundamente mi atención, es la rapidez con que se difunden las noticias medicas en todos los medios; periódicos, internet. Y para todo el que sepa leer.
Esto no hay libro que lo soporte
Bibliografia
FDA approves the first treatment directed at the underlying biology of Alzheimer’s disease, which is expected to reduce the clinical decline of patients with this disease: https://t.co/xEVvn7V2Fipic.twitter.com/OaADO8DkZB — U.S. FDA (@US_FDA) June 7, 2021
Todas las enfermedades neurodegenerativas se caracterizan por el acumulo de proteínas en lugares múltiples que bloquean de forma progresivas , no solo funciones nerviosas sino múltiples estructuras de nuestra economía
Células de levaduras en depósitos de Amiloide
Es bastante probable que las enfermedades degenerativas y concretamente las neurodegenerativas, tengan un patrón microbiano y desde aquí desarrollen una respuesta inmunitaria que sea responsable de la destrucción cerebral
El Parkinson que se caracteriza por perdida de las estructuras dopaminérgicas donde se depositan las proteínas cuerpos de Lewy.
Los cuerpos de Lewy son depósitos anormales de proteína llamada alfa-sinucleína. Que son propias en la demencia producidas por estosl deposito de Pero saben que otras enfermedades, como el mal de Parkinson, también involucran la acumulación de esta proteína.
Demencias con cuerpos de Lewy se presentan por ahora en :
Demencia con cuerpos de Lewy y demencia por enfermedad de Parkinson. Ambos tipos causan los mismos cambios en el cerebro, y, con el tiempo, provocan los mismos síntomas. La principal diferencia es cuándo los síntomas cognitivos (del pensamiento) y del movimiento comienzan.
También la demencia con cuerpos de Lewy causa problemas con la habilidad de pensar similar a la enfermedad de Alzheimer . Posteriormente, causa otros problemas, como síntomas de movimiento, alucinaciones visuales y ciertos problemas del sueño. También causa más problemas con las actividades mentales que con la memoria.
La demencia por enfermedad de Parkinson comienza como un trastorno del movimiento. Primero causa los síntomas de la enfermedad de Parkinson, movimientos lentos, rigidez muscular, temblor y caminar arrastrando los pies. Más adelante, causa demencia.
La demencia con cuerpos de Lewy ocurre cuando se acumulan cuerpos de Lewy en partes del cerebro que controlan la memoria, el pensamiento y el movimiento.
Otras dos enfermedades neurodegenerativas tienen un acumulo anormal de proteínas, que actúan como macrofagos El Alzheimer (AL) es una enfermedad neurodegenerativa, y la causa principal y más conocida en enfermedades demenciales, cuya característica principal es la pérdida de memoria. Su relación con la proteína amiloide y tau es indiscutible. Su acumulo en el lóbulo temporal entre otras zonas del cerebro mutila la funciones cognoscitiva y produce demencia. La Esclerosis Múltiple, (EM) también es una enfermedad neurodegenerativa, y afecta a los nervios, destruye la mielina que es su aislante Los nervios están envueltos en mielina que los aísla y permite su función. La beta-amiloide sería capaz de luchar contra la reacción inflamatoria autoinmune que causa la EM, la proteína beta-amiloide y su precursor se encuentran en las lesiones de la EM .
La administración de proteínas beta-amiloide fuera del cerebro, concretamente se inyectó en el vientre de los ratones y no en el cerebro. Aunque los sistemas inmunes de estos ratones estaban preparados para atacar al aislante nervioso o mielina, con la administración de la beta-amiloide se produjo todo lo contrario.
Así lo expresa Steinman: “Esta es la primera vez que la beta amiloide demuestra tener propiedades antiinflamatorias” En definitiva, la beta-amiloide fuera del sistema nervioso central tendría propiedades como antiinflamatorio, todo lo contrario a lo que produce cuando se encuentra dentro del mismo cerebro o de los nervios. El juego inflamatorio en enfermedades degenerativas, llegan siempre a las misma situaciones, un problema de inflamación y reparación complejo e imbricado. No es la primera vez que se analizan las propiedades de la beta amiloide, El Hospital General de Massachusetts (MGH,), evidencio que la proteína beta-amiloide se deposita en forma de placas en el cerebro de pacientes con enfermedad de AL y se la considera una parte normal del sistema inmune innato, primera línea de defensa del cuerpo contra la infección. Su estudio, publicado en ‘Science Translational Medicine’, concluye que la expresión de beta-amiloide humana resulta protectora contra las infecciones potencialmente letales en ratones, en el C elegans y en células cerebrales humanas en cultivo.
Se admite que la neurodegeneración en la enfermedad de AL es causada por el comportamiento anormal de moléculas de beta-amiloide, que son conocidas por reunirse en resistentes estructuras de fibrillas llamadas placas amiloides en el cerebro de los pacientes En 2010 , Moir y Rudolph Tanzi, director del MGH-MIND y concluyeron que la beta-amiloide tenía muchas de las cualidades de un péptido antimicrobiano (AMP) y se trata de una pequeña proteína innata del sistema inmune que protege contra una amplia gama de patógenos. En ese estudio se compararon formas sintéticas de A-beta con un conocido AMP llamado LL-37 y se encontró que la beta inhibe el crecimiento de varios patógenos importantes, a veces igual de bien o mejor que LL-37. La beta amiloide de los cerebros de los pacientes de AL también suprimió el crecimiento del hongo ‘Candida’ cultivado para esa investigación y, posteriormente, otros grupos han documentado la acción de A-beta sintético contra los virus de influenza y herpes. En este nuevo trabajo, los investigadores encontraron que los ratones transgénicos que expresan A-beta humano sobrevivieron significativamente más tiempo después de inducir la infección por ‘Salmonella’ en sus cerebros frente a los ratones sin alteración genética. Los ratones que carecen de la proteína precursora de amiloide murieron incluso más rápidamente. La expresión de la beta transgénica parece proteger a los gusanos ‘C.elegans’ de cualquier infección por ‘Candida’ o ‘Salmonella’. Del mismo modo, la expresión de beta humana protege las células neuronales cultivadas de ‘Candida’. De hecho, la beta amiloide humano expresado por células vivas parece ser mil veces más potente contra la infección que el beta amiloide sintético utilizado en estudios previos. Esa superioridad parece referirse a propiedades de beta amiloides que se han considerado parte de la patología en la enfermedad AL, la propensión de moléculas pequeñas a combinarse en lo que se denominan oligómeros y luego se agregan en placas de beta-amiloide.
Moir añade: “Nuestros resultados plantean la intrigante posibilidad de que puede surgir la patología de AL, cuando el cerebro se percibe a sí mismo como bajo el ataque de los patógenos invasores, aunque se necesitan estudios adicionales. No parece probable que las vías inflamatorias del sistema inmune innato puedan ser posibles dianas de tratamiento. Si se validan, nuestros datos también justifican la necesidad de tener precaución con terapias dirigidas a la eliminación total de las placas de beta-amiloide. Las terapias basadas en la disminución de amiloides, pero no en la eliminación de beta amiloide en el cerebro podría ser una estrategia mejor”.
Un nuevo estudio de la Universidad de Florida, en Estados Unidos, establece, por primera vez, un vínculo entre especies específicas de bacterias y manifestaciones físicas de enfermedades neurodegenerativas.
Investigaciones recientes sugieren que las personas con estas enfermedades presentan cambios en la composición bacteriana de su tracto digestivo.
Una nueva investigación publicada en ‘PLOS Pathogens’ establece, por primera vez, un vínculo entre especies específicas de bacterias y manifestaciones físicas de enfermedades neurodegenerativas.
En este estudio, quiere demostrar que especies específicas de bacterias desempeñan un papel en el desarrollo de estas enfermedades», Daniel Czyz,
Algunas otras bacterias producen compuestos que contrarrestan estas bacterias ‘malas’. Estudios recientes han demostrado que los pacientes con la enfermedad de Parkinson y de Alzheimer son deficientes en estas bacterias ‘buenas’, por lo que nuestros hallazgos pueden ayudar a explicar esa conexión y abrir un área de estudio futuro», añade.
Todas las enfermedades neurodegenerativas tienen su origen en problemas en el manejo de las proteínas en el organismo. Si las proteínas están mal plegadas, se acumulan en los tejidos. Estos agregados de proteínas, como los llaman los científicos, interfieren en el funcionamiento de las células y provocan trastornos neurodegenerativos.
Czyz y sus coautores querían saber si la introducción de ciertas bacterias en los gusanos ‘C. elegans’ iría seguida de la agregación de proteínas en los tejidos de los gusanos.
» Con una forma de marcar los agregados para que brillen en verde bajo el microscopio, se vio que los gusanos colonizados por ciertas especies de bacterias se iluminaban con agregados que eran tóxicos para los tejidos, mientras que los colonizados por las bacterias de control no lo hacían. Esto ocurría no sólo en los tejidos intestinales, donde están las bacterias, sino en todo el cuerpo de los gusanos, en sus músculos, nervios e incluso órganos reproductores».
Sorprendentemente, las crías de los gusanos afectados también mostraron un aumento de la agregación de proteínas, a pesar de que estas crías nunca se encontraron con las bacterias originalmente asociadas a la enfermedad.
«Esto sugiere que estas bacterias generan algún tipo de señal que puede transmitirse a la siguiente generación»,
Czyz. Afirma que los gusanos colonizados por las bacterias «malas» también perdieron movilidad, un síntoma común de las enfermedades neurodegenerativas.
«Un gusano sano se mueve rodando y dando vueltas. Cuando se coge un gusano sano, se desplaza rodando por el pico, un sencillo dispositivo que se utiliza para manipular estos diminutos animales. Pero los gusanos con la bacteria mala no podían hacerlo debido a la aparición de agregados de proteínas tóxicas», explica Walker, que desarrolló este método de evaluación.
Los gusanos son muy delicados, así que se necesita una herramienta que no los dañe. Además, son transparentes y tienen un plan corporal sencillo. Estudios como este son posibles porque estos gusanos se alimentan normalmente de bacterias.
«Los gusanos sólo miden un milímetro de largo y cada uno tiene exactamente 959 células . Pero, en muchos aspectos, se parecen mucho a los humanos: tienen intestinos, músculos y nervios, pero en lugar de estar compuestos por miles de millones de células, cada órgano es sólo unas pocas células.
Su pequeño tamaño nos permite hacer experimentos de forma mucho más controlada y responder a preguntas importantes que podremos aplicar en futuros experimentos con organismos superiores y, eventualmente, con personas».
En la actualidad, el laboratorio de Czyz está probando cientos de cepas de bacterias presentes en el intestino humano para ver cómo afectan a la agregación de proteínas en ‘C. elegans’. El grupo también investiga cómo las bacterias asociadas a la neurodegeneración provocan el mal plegamiento de las proteínas a nivel molecular.
Czyz también está interesado en las posibles conexiones entre las bacterias resistentes a los antibióticos y el mal plegamiento de las proteínas.
Lo que se deduce de estos estudios, es que el deposito de proteínas mal plegadas, se hace sobre gérmenes previos y no al revés.
«Casi todas las bacterias que encontramos asociadas al mal plegamiento de las proteínas están también asociadas a infecciones resistentes a los antibióticos en las personas.
Varios autores sobre todo Carrasco y Moir, han repetido de forma machacona, como los gérmenes están presentes en las enfermedades neurodegenerativas y que los depositos de proteínas plegadas que son macrófagos es un intento del organismo por eliminar estos gérmenes.
El desequilibrio empieza en la microbiota intestinal, se sigue de la inflamación expresada por macrófagos, y la aparición de enfermedades neurodegenerativas
Gérmenes en el Alzheimer
BIBLIOGRAFÍA
Sociedad Española de Medicina de Familia y Comunitaria. Demencias desde la Atención Primaria. Barcelona: semFYC; 2005. 2. Grupo de trabajo de la Guía de Práctica Clínica sobre la atención integral a las personas con enfermedad de Alzheimer y otras demencias. Guía de Práctica Clínica sobre la atención integral a las personas con enfermedad de Alzheimer y otras demencias. Plan de Calidad para el Sistema Nacional de Salud del Ministerio de Sanidad, Política Social e Igualdad. Agència d´Informació, Avaluació i Qualitat en Salut de Cataluña; 2010. Guías de Práctica Clínica en el SNS: AIAQS Núm. 2009/07. http://portal.guiasalud.es/web/guest/home 3. Hort J, O’Brien JT, Gainotti G, Pirttila T, Popescu BO, Rektorova I, Sorbi Sand and Scheltensh P. EFNS guidelines for the diagnosis and management of Alzheimer´s disease. Eur J Neurol 2010; 17: 1236-1248. 4. National Institute for Health and Clinical Excellence (NICE). Clinical guideline 42. Dementia: supporting people with dementia and their carers in health and social care. November 2006 (last modified: March 2011). http://www.nice.org.uk/CG42 5. Feldman HH, Jacova C, Robillard A, et al. Diagnosis and treatment of dementia: 2. Diagnosis. CMAJ 2008;178: 825-36. http://www.ecmaj.ca/cgi/content/abstract/178/7/825 6. Knopman DS, DeKosky ST, Cummings JL, et al. Practice parameter: diagnosis of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001: 56:1143-1153. http://www.neurology.org/content/56/9/1143.full.pdf+html 7. Olazaran J, Alteración cognitiva leve en la práctica clínica. Med Clin. 2011; 414-418. 8. Albert MS et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7: 270-9. 9. Robles A, Del Ser T, Alom J, Peña-Casanova J y Grupo Asesor del Grupo de Neurología de la Conducta y Demencias de la Sociedad Española de Neurología. Propuesta de criterios para el diagnóstico clínico del deterioro cognitivo ligero, la demencia y la enfermedad de Alzheimer. Neurología. 2002; 17(2): 17-32. 2 10. Chertkow H, Massoud F, Nasreddine Z, et al. Diagnosis and treatment of dementia: 3. Mild cognitive impairment and cognitive impairment without dementia. CMAJ. 2008;178:1273-85. http://www.cmaj.ca/cgi/content/abstract/178/10/1273 11. Petersen RC, Stevens JC, Ganguli M et al. Practice parameter: Early detection of dementia: Mild cognitive impairment (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001; 56: 1133-1142. http://www.neurology.org/content/56/9/1133.full.pdf+html 12. Petersen RC, Doody R, Kurz A, et al. Current concepts in mild cognitive impairment. Arch Neurol. 2001; 58:1985–1992 13. Contador I, Fernández-Calvo B, Ramos F, Tapias-Merino E, Bermejo-Pareja F. El cribado de la demencia en atención primaria. Revisión crítica. Rev Neurol. 2010; 51: 677-86. http://www.revneurol.com/sec/resumen.php?id=2010453# 14. Villarejo A, Puertas-Martín V. Utilidad de los test breves en el cribado de demencia. Neurología. 2011; 26 (7): 425-433 15. De Hoyos Alonso, Tapias Merino E, García de Blas F. Demencia. En: Los principales problemas de salud. AMF 2012; 8(9): 484-495. 16. Olazarán. J. Bermejo F: ¿Puede diagnosticarse la demencia en Atención Primaria? Aten Primaria. 2011; 43 (7): 377-384 17. Simmons B, Hartmann B, Dejoseph D. Evaluation of suspected dementia.. Am Fam Physician. 2011 Oct 15;84(8):895-902. 18. Riu S y Martínez A. Síndrome confusional agudo en el mayor. AMF. 2008; 4(4):216-21 19. American Geriatrics Society Updated Beers criteria for potentially Inappropriate medications use in older adults. American Geriatrics Society 2012 Beers Criteria Update Expert panel. JAGS 2012 http://www.americangeriatrics.org/files/documents/beers/2012BeersCriteria_JAGS.pdf 20. Riu S, Martínez A y Baena D. Fármacos que pueden alterar el estado cognitivo en el anciano. FMC.2009;16(5):
Alzheimer y Robert Moir « Enriquerubio.net
Luis Carrasco,. Catedrático de Microbiología de la UAM,
.Un nuevo mecanismo tóxico que bloquea cualquier reacción celular que use ácidos nucleicos originaría la muerte de las neuronas motoras en pacientes con ELA.
La esclerosis lateral amiotrófica (ELA), la “enfermedad de Lou Gehrig”, es una enfermedad neurodegenerativa progresiva que afecta a las células nerviosas del cerebro y de la médula espinal. Las neuronas motoras van del cerebro a la médula espinal y de la médula espinal a los músculos de todo el cuerpo.
A-mio-trófica proviene del griego. «A» significa sin o carente. «Mio» se refiere a los músculos, y «trófica» significa alimentación: «Sin alimentación a los músculos». Cuando un músculo no es alimentado, se «atrofia» o se desgasta. «Lateral» identifica las áreas de la médula espinal donde se localizan partes de las células nerviosas que dan señales y controlan los músculos. A medida que esta área se va degenerando, produce la cicatrización o el endurecimiento («esclerosis») en la región.
A medida que las neuronas motoras se van degenerando, dejan de enviar impulsos a las fibras musculares que normalmente resultan en el movimiento muscular. Los primeros síntomas de la ELA a menudo incluyen una mayor debilidad muscular, especialmente en brazos y piernas, en el habla, en la acción de tragar o en la respiración. Cuando los músculos dejan de recibir los mensajes de las neuronas motoras que requieren para funcionar, se empiezan a atrofiar (se vuelven más pequeños). Las extremidades se empiezan a ver más «delgadas», a medida que se atrofia el tejido muscular.
Astas anteriores de la médula espinal con atrofia, degeneración y cromatolisis de neuronas
Desmielinización de cordones laterales de medula espinal
FACTORES de riesgo EN LA esclerosis lateral amiotrófica:
Factor hereditario. Entre el 5 y el 10 % de las personas con esclerosis lateral amiotrófica la heredaron (esclerosis lateral amiotrófica familiar). La mayoría de los hijos de personas con esclerosis lateral amiotrófica familiar tienen un 50 % de probabilidades de desarrollar la enfermedad.
La edad. El riesgo de esclerosis lateral amiotrófica aumenta con la edad, y es más común entre los 40 y mediados de los 60 años.
Sexo. Antes de los 65 años, la esclerosis lateral amiotrófica es un poco más común en hombres que en mujeres. Esta diferencia de sexo desaparece después de los 70 años.
La genética. Algunos estudios que examinan todo el genoma humano encontraron muchas similitudes en las variaciones genéticas de las personas con esclerosis lateral amiotrófica familiar y algunas personas con esclerosis lateral amiotrófica no hereditaria. Estas variaciones genéticas podrían hacer que las personas sean más susceptibles a la esclerosis lateral amiotrófica.
Se trata de la tercera enfermedad neurodegenerativa con mayor incidencia –después de la demencia y el Parkinson-, y de elevada mortalidad. Se calcula que cada año aparecen unos 2-5 casos por cada 100.000 habitantes en el mundo. En España, se producen tres nuevos pacientes diagnosticados cada día.
Actualmente, carece de tratamiento y su causa es aún una incógnita. No obstante, se sabe que en el 10% de los afectados hay un importante componente genético, que hace que la enfermedad aparezca en varios miembros de una familia.
En casi la mitad de estos casos de ELA familiar el origen está en un gen llamado C9ORF72 en el cual, sus mutaciones tienen un papel destacable.
“A principios de 2011 se descubrió que la mutación más frecuente en pacientes de ELA se encuentra en este gen. Pocos años después, se confirma que esta mutación acaba produciendo unos pequeños péptidos que son tóxicos, pero se desconocía el mecanismo que conducía a ello y que ahora se ha identificado en el CNIO»,
En la mutación del C9ORF72 de los pacientes con ELA existe una pequeña secuencia repetida de ADN que, en personas no enfermas suele ser de unas 8-10 copias, y que en estos pacientes está expandida hasta ciento de veces. “Esta expansión de repeticiones se traducen en proteínas que generan unos pequeños péptidos que son tóxicos, un fenómeno que, por otra parte, se ha observado en otros contextos de la naturaleza humana y para el que hemos desarrollado un modelo que los conecta a todos y explica así estos problemas tan generalizados”.
El mecanismo tóxico identificado y que está asociado a un gen concreto, el C9ORF72, “no se descarta la posibilidad que otras mutaciones relacionadas con ELA estén actuando de manera similar, es decir, bloqueando el ADN y ARN de las neuronas motoras”.
Aprender a aliviar la toxicidad de estos péptidos puede ser útil también para abordar los casos de ELA no asociados a C9ORF72, lo que englobaría a la enfermedad en su conjunto.
“la gran mayoría de las mutaciones halladas en pacientes de ELA son en proteínas que se unen a ARN, y generalmente lo que hacen estas mutaciones es impedir la unión de estas proteínas al ARN».
Además, las células de estos pacientes también tienen problemas muy generales con sus ácidos nucleicos, por lo que «pensamos que, aunque las mutaciones en C9ORF72 afectan a solo una parte de los pacientes de ELA, el mecanismo subyacente a la toxicidad de las neuronas puede que no sea muy diferente, en lo fundamental, a lo que pasa en el resto de los casos de esta enfermedad”.
“
Bibliografía
Crawford TO. From enigmatic to problematic. The new molecular genetics of childhood spinal muscular atrophy. Neurology 46: 335-340, 1996.
Ikeda M et al: variable clinical symptoms in familiar amyotrofic lateral sclerosis with a novel point in the Cu/Zn superoxide dismutase gene. Neurology 45: 2038-2041, 1995.
Rosen DR et al: Mutations in Cu/Zn superoxide dismutase gene are associated with familiar amyotrofic lateral sclerosis. Nature 362: 59, 1993.
Siddique T et al: Linkage analysis in familial amyotrofic lateral sclerosis, Neurology 39: 919-926, 1989.
Veltema AN, Ross RAC, and Bruyn GW: Autosomal dominant adult amyotrofic lateral sclerosis, J Neurol Sci 97: 93-115, 1990.
LaSpada AR, et al: Androgen receptor gene mutations in X linked spinal and bulbar muscular atrophy, Nature 352: 77-79, 1991.
Rohstein JD, et al: Abnormal excitatory amino acid metabolism in amyotrofic lateral sclerosis, Ann Neurol 28: 18-25, 1990.
Younger DS, et al: Motor neuron disease and ALS: Relation of high CSF protein content to paraproteinemia and clinical syndromes, Neurology 40: 595-599, 1990.
Williams C, et al: Degeneration of spinocerebellar neurons in amyotrofic lateral sclerosis, Ann Neurol 27: 215-225, 1990.
Brownell B, Oppenheimer DR, and Hughes TJ: The central nervous system in motor neuron disease, J Neurol Neurosurgery Psychiatry 33: 338-357, 1970.
Alvarado F Rodriguez V Esclerosis Lateral amiotrófica. Casos familiares en 6 hermanos. En preparación. Archivos del Departamento de Patología. Hospital México.
Cotran RS, Kumar V, Collins T. Robbins. Patologia estructural y funcional. 6ª ed. Madrid.- McGraw-Hill/Interamericana, 2000.
Neuropathology The diagnostic Approach. St Louis: Mosby, 1977. Raquel Serrano
Mis enfermos, tienen más de 70 años y hasta 96 y el mal más corriente es de múltiples infartos cerebrales, diabetes, demencias, y caidas y sobre todo dolores articulares y dominandolo todo; están tristes, no duermen bien, no tienen ganas de nada y están llenos de dolores, cansancio extremo y tienden a la inmovilidad.
El trato que se les da es siempre exquisito y la medicación lo mas ambiciosa posible para cubrir todos los aspectos de su patología. Y si que mejoran, recuperan las parálisis o parte de ella, se mejora la cifra de azúcar en sangre, duermen algo mejor y les quitamos los dolores. Pero con frecuencia los resultados no son buenos y con frecuencia recidivan los brotes.
Cada vez vivimos más. Yo recuerdo mis tiempos de Neurocirujano en Sevilla y la edad media era sensiblemente menor, de forma que si algún paciente con mas de 60 años, tenía un tumor cerebral o un hematoma o una patología importante en el cerebro, “nos pensábamos en operarlo”, porque los resultados no eran buenos.
Hoy no es infrecuente que operemos a un señor con 90 años y además quede aceptablemente bien.
Es verdad que la técnica, las unidades de cuidados intensivos y todo un arsenal de medicina ha permitido esto.
Pero yo hablo del señor enfermo y su familia que están metidos en la tristeza, y que le dan vuelta a todo y que cualquier cosa es en su opinión insufrible. Un paciente que ha tenido un cuadro de insuficiencia vascular cerebral ,o una caída, por ejemplo, y le produce un defecto de la motilidad de uno de sus hemicuerpos, por muy bien que quede, no estará nunca como antes de sufrir el ictus, o de la caída. Forzosamente aquel enfermo queda peor que antes y no solo físicamente sino siempre con quejas, con matices poco importantes que le amargan la vida. Porque ser feliz no es un estado sublime que nos acerca a lo exquisito. Ser feliz es no tener dolor ni malestar interior. Así de sencillo y así de terriblemente difícil.
Un psiquiatra conocido, dulce y sabio al que repetidamente he oído hablando del poder de la palabra y como él consigue mejorar el ánimo de los pacientes, jóvenes y viejos con simple consejos y lo que más me sorprendio, es que los pacientes le hacen caso.
Esto no es distinto de lo que vende determinada literatura, con fármacos mágicos con los que obtienen la salud fácilmente, y convierten en bienestar unos simples consejos.
Cuando hablo con mis colegas a los que puedo abordar, y les habló del tema, ellos afirman que cuando un enfermo, afectado de un grave proceso logra sobreponerse y es capaz de superarse y de despreocuparse de sus dolores, insomnio, insatisfacción y ganas de no salir a la calle y estar siempre en la cama. Ellos me contestan que si, que es posible y además me parece que tienen cara de decir la verdad.
Yo me confundo, pues esto no ocurre ni una sola vez en mis pacientes. si no duermen, y no les doy algo que lo induzca al sueño, que le mejore el dolor y algún antidepresivo. Les aseguro que se quedan sin dormir, se levanta cansado, no tienen ganas de leer, ni ver TV, ni salir a la calle, ni casi de nada.
Esto que parece un estado de desesperación ante lo poco efectiva de la ayuda que estamos prestando a los enfermos con enfermedades cerebrales y distimias, que son muchos , y que estamos intentando convencerlos que la ayuda viene de ellos, que se puede autoayudar. Nunca lo he creído. Es posible que lo consigan algunos profesionales muy ensayados, pero a mi cuesta creerlo.
Hace falta conocer que le pasa a nuestro cerebro, porque se lesiona tan fácilmente y ello no le permite no solo la alegría, sino le induce a la tristeza.
Estudios sobre las enfermedades demenciales sobre todo de la enfermedad de Alzheimer, demuestran que el cerebro de estos enfermos llenos de detritus, placas seniles, por acúmulo de proteínas beta amiloidotica y la Tau intracelular, que representan un fenómeno inflamatorio, pero como reacción a una agresion previa, que siempre creo debida a un germen o a varios de ellos. Esto difícilmente se va a curar con la palabra por muy amorosas que estas sean.
Lo que si parece logico es la posibilidad que algún tipo de germen , o varios de ellos, sean los iniciante del proceso y una reacción de nuestro cerebro y de nuestro organismo en general llene al cerebro de basura que dificulta la conducción neuronal.
El otro factor es la frecuencia con que se reagudiza su enfermedad, con un disgusto, con un cambio de hábitat, con una intervención o con un cambio de medicación y sobre todo con una caída. Entonces, todo se torna dramático, pasan de un estado de alucinaciones, agitación y desespero a otros de una indiferencia, quietud y hasta coma. Todo ello parece un estado terminal que no solo asusta a los familiares, sino a médicos y enfermeras que pese a su frecuencia no se habitúan a este delirio del anciano. Pero en la mayoría de los casos se controla con medicamentos que inhiben la DOPA, aunque si la dosis de medicación no es la adecuada, aparece un cuadro extrapiramidal, también dramático.
Tras una medicación acertada y el paso de unos días el paciente vuelve a su estado previo de una manera sorprendente en un alto porcentaje de casos.
Pretendo decir, que cualquier accidente, precipita o desencadena el asiento de gérmenes y la reacción inflamatoria consiguiente
Un germen, una reacción inflamatoria que tiende a la cronicidad, un deterioro y en ocasiones el proceso inflamatorio.
Esta organicidad de la evolución condiciona su alteración psiquica que con frecuencia conduce al final.
De forma que yo no me niego a la terapia de la palabra y al cariño que hay que dar a cualquier paciente, sino lo que afirmo es que la palabra ni el cariño tienen fuerza para controlar el proceso y si el conocimiento y la medicación adecuada.
La psicoterapia tiene sus indicaciones el amor y el máximo de los cuidados al paciente hay que practicarlo siempre como principio, pero las enfermedades son orgánicas y hay que tratarlas como tales, el cuadro psíquico que se les añade es también por deterioro de su cerebro y rara vez porque el enfermo presida su deterioro.
Pero la palabra cuando la organicidad es la responsable del daño, no sirve para casi nada.
Este “el golpe”, desencadena una serie de fenómenos hasta entonces ocultos, que terminan lesionando o matando al enfermo.
Las caídas son una causa de mortalidad en los pacientes añosos. Esto se comporta como las enfermedades neurodegenerativas, las cuales, no las tenemos aun por la mano.
Investigar el porque del deposito de proteínas. Matar los gérmenes antes que actúen y mientras tanto todo el cariño del mundo y sobre todo evitar las caidas
EL SÍNDROME DE GUILLAN BARRE UNA ENFERMEDAD NEURODEGENERATIVA
Histologia en Guillan Barre
La cronicidad que es la característica común de estas enfermedades. En este síndrome existe poco tiempo entre el contagio y la clínica, no obstante, se comporta como lo suelen hacer las neurodegenerativas:
Asociación de gérmenes, probablemente varios como en otras enfermedades de este tipo, y la desmielinización, la hacen una enfermedad infecciosa e inflamatoria.
Guillain-Barré es un síndrome y la presentación clínica aunque variada, la parálisis flácida aguda es el síntoma dominante aunque en territorios dispares.
La mayoría de los pacientes se presentan con antecedentes de enfermedad, con mayor frecuencia infección del tracto respiratorio superior, antes del inicio de la debilidad motora progresiva.
Varios microorganismos se han asociado con el síndrome de Guillain-Barré, en particular Campylobacter jejuni , virus Zika y, en 2020, el síndrome respiratorio agudo severo coronavirus 2.
En C jejuni relacionado con el síndrome de Guillain-Barré, existe buena evidencia que respalda un proceso inmunológico mediado por autoanticuerpos que se desencadena por el mimetismo molecular entre los componentes estructurales de los nervios periféricos y el o lo que se sostienen en la actualidad por muchos autores, sobre los gérmenes que infectan, se acumulan macrófagos que desmielinizan las estructuras nerviosas periféricas.
Hacer un diagnóstico del llamado síndrome de Guillain-Barré clásico es sencillo; sin embargo, los criterios de diagnóstico existentes tienen limitaciones y pueden dar lugar a que se pasen por alto algunas variantes del síndrome
Lesion axonal en Guillan Barre
Diagnostico.
Clasicamente la disociación albumino citológica definia el diagnostico. En la actualidad, y pese a su frecuencia, la forma solapada de algunos casos, dificulta el diagnostico. La dificultad respiratoria, con su frecuente incidencia confirma el caso. El síndrome de Guillain-Barré puede ser difícil de diagnosticar en las primeras etapas. Sus signos y síntomas son similares a los de otros trastornos neurológicos y pueden variar de persona a persona.
El síndrome de Guillain-Barré no tiene cura. Pero dos tipos de tratamientos pueden acelerar la recuperación y reducir la gravedad de la enfermedad:
Intercambio de plasma (plasmaféresis). La porción líquida de parte de la sangre (plasma) se extrae y se separa de las células sanguíneas. Luego, las células sanguíneas se vuelven a colocar en el cuerpo, el cual produce más plasma para compensar lo que se extrajo. La plasmaféresis puede funcionar liberando al plasma de ciertos anticuerpos que contribuyen al ataque del sistema inmunitario a los nervios periféricos.
Terapia de inmunoglobulina. La inmunoglobulina que contiene anticuerpos sanos de donantes de sangre se administra a través de una vena (por vía endovenosa). Las dosis altas de inmunoglobulina pueden bloquear los anticuerpos perjudiciales que podrían contribuir al síndrome de Guillain-Barré.
. A la mayoría de los pacientes con síndrome de Guillain-Barré les va bien con la inmunoterapia, pero una proporción sustancial queda con discapacidad y puede ocurrir la muerte. Los resultados del Estudio Internacional de Resultados del Síndrome de Guillain-Barré sugieren que existen variaciones geográficas en el síndrome de Guillain-Barré, incluido el acceso insuficiente a la inmunoterapia en los países de bajos ingresos.
Estos tratamientos son igualmente eficaces. Combinarlos o administrar un tratamiento tras otro no es más eficaz que usar cualquiera de los dos métodos por separado.
También beneficia estos paciente.
Aliviar el dolor, que puede ser intenso
Prevenir los coágulos sanguíneos, que se pueden desarrollar mientras estás inmóvil
Las personas con el síndrome de Guillain-Barré necesitan ayuda física y fisioterapia antes y durante la recuperación. Movimiento de los brazos y las piernas por parte de las personas encargadas del cuidado antes de la recuperación, para ayudar a mantener los músculos flexibles y fuertes
Fisioterapia durante la recuperación para ayudarte a lidiar con la fatiga y recuperar la fuerza y el movimiento adecuado
Entrenamiento con dispositivos de adaptación, como una silla de ruedas o aparatos ortopédicos, para brindarte movilidad y habilidades de cuidado personal
Recuperación.
Si bien a muchas personas les llevan meses e incluso años recuperarse, la mayor parte de las personas con síndrome de Guillain-Barré siguen esta cronología general:
Después de los primeros signos y síntomas, la afección tiende a empeorar progresivamente durante aproximadamente dos semanas
Los síntomas llegan a una meseta en cuatro semanas
Comienza la recuperación, que suele durar de seis a 12 meses, aunque para algunas personas puede durar hasta tres años
Entre los adultos que se recuperan del síndrome de Guillain-Barré:
Alrededor del 80 % puede caminar independientemente seis meses después del diagnóstico
Alrededor del 60 % recupera completamente la fuerza motora un año después del diagnóstico
Alrededor del 5 % al 10% tiene una recuperación muy retrasada e incompleta
Los niños, que rara vez presentan el síndrome de Guillain-Barré, por lo general se recuperan más completamente que los adultos.
Actualmente se están realizando ensayos clínicos para investigar algunos de los posibles candidatos terapéuticos, incluidos los inhibidores del complemento, que, junto con los datos emergentes de grandes estudios colaborativos internacionales sobre el síndrome, contribuirán sustancialmente a comprender las múltiples facetas de esta enfermedad y desarrollar terapias eficaces para modificar la enfermedad que puedan limitar la extensión de la lesión nerviosa
Referencias
Sejvar JJ Baughman AL Wise M Morgan OW
Incidencia poblacional del síndrome de Guillain-Barré: una revisión sistemática y un metanálisis.
El oncólogo barcelonés Josep Baselga, fue director médico del Memorial Sloan-Kettering Cancer Center de Nueva York y el primer director del Instituto De Oncología del Vall d’Hebron
Fuimos compañeros en el Valle de Hebron y a todos nos sorprendia su sabiduría y su capacidad de innovar.
El tratamiento de los tumores de mama de manera individualizada proporciono unos explendidos resultados, y claro los Americanos lo reclamaron y allí desarrollo una investigación solida.
Esta maldita y afortunadamente rara enfermedad, no sé cómo lo contagio.
Amigo que descanses en la paz de Dios
El oncólogo barcelonés Josep Baselga, actual director del área de Investigación y Desarrollo (I+D) para oncología de la compañía AstraZeneca, ha muerto este domingo a los 61 años, según ha hecho público el Hospital Vall d’Hebron de Barcelona en un comunicado. ‘La Vanguardia’ ha informado de que la causa de su fallecimiento ha sido la enfermedad de Creutzfeldt-Jakob.
Afincado Estados Unidos, fue director médico del Memorial Sloan-Kettering Cancer Center de Nueva York y el primer director del Instituto De Oncología del Vall d’Hebron. Tras estudiar medicina en la Universidad Autónoma de Barcelona, inició su formación en Medicina Interna en el Hospital Universitario Vall d’Hebron, que completó en el Hospital Kings County de Brooklin y posteriormente en el área de oncología del Memorial Sloan Kettering de Nueva York.
Baselga regresó a España en 1996 como profesor titular de la UAB y coordinador y jefe del servicio de oncología médica de Vall d’Hebrón, donde, bajo su dirección, creó un departamento de oncología, que fue pionero y referente internacional, al integrar la asistencia a pacientes con un programa de investigación básica y clínica para trasladar a los enfermos de cáncer, con la mayor rapidez posible, los avances que se realizaban en el laboratorio.
Por iniciativa suya, creó el VHIO en 2006 y lo dirigió hasta su marcha a Estados Unidos, donde en 2010 se trasladó a Boston para dirigir la división de oncología, con más de cien investigadores, del Hospital General de Massachusetts, aunque lo compaginaba manteniendo la actividad científica en Vall d’Hebron.
Entre los años 2013 y 2018 asumió la dirección médica del Memorial Sloan Kettering Cancer Center de Nueva York y desde 2019 era el director mundial de I+D del Área de Oncología de la farmacéutica británica AstraZeneca.
Durante su trayectoria, recibió premios como el Rosenthal Family Foundation, el Pezcoller Award, el Premi Jaime I, la Medalla Trueta y el Premi Internacional de Catalunya.
Su enfermedad fue descrita en 1920 por el neurólogo alemán Hans Gerhard Creutzfeldt y poco después por su homólogo Alfons Maria Jakob. Una variante de la enfermedad de Creutzfeldt-Jakob, la llamada enfermedad de las vacas locas, causó un grave problema de salud pública a finales del siglo XX. Causó la muerte prematura de 170 personas en el Reino Unido y 50 en el resto del mundo que habían ingerido priones infecciosos procedentes del cerebro de animales enfermos.
La enfermedad de Creutzfeldt-Jakob, que ha causado la muerte del oncólogo Josep Baselga, es una rara enfermedad neurológica degenerativa de evolución rápida.
Tiene una incidencia de un caso por millón de personas al año, según datos del Centro de Prevención y Control de Enfermedades de EE.UU. (CDC). Puede afectar tanto a hombres como a mujeres. Suele manifestarse entre los 60 y los 70 años de edad.
Se caracteriza por un rápido deterioro del tejido cerebral causado por un prión, una proteína defectuosa que se extiende como una infección. Forma parte del grupo de las encefalopatías espongiformes, porque el cerebro se degrada de una manera que le da una apariencia de esponja.
Los síntomas iniciales incluyen dificultades de memoria, problemas de coordinación y alteraciones de comportamiento. A medida que la enfermedad progresa, los pacientes suelen experimentar movimientos involuntarios, trastornos de personalidad, alteraciones del estado de ánimo y demencia.
Suelen transcurrir entre tres y seis meses entre el inicio de síntomas y la muerte, aunque algunos casos tienen supervivencias algo más largas. No existe ningún tratamiento para frenar la progresión de la enfermedad: es mortal en el 100% de los casos.
Alrededor de un 10% de los casos tiene un componente hereditario conocido. La gran mayoría son casos esporádicos cuya causa se desconoce.
HISTORIA DE LA ENFERMEDAD DE PARKINSON DURANTE LOS DOS ÚLTIMOS SIGLOS
“Essay on the shaking palsy “,es el nombre que le dio James Parkinson a esta enfermedad, Y pese a que es muy antigua, este nombre persiste . Fue publicado en 1817, y su descripcion fue y es magistral1
Esta enfermedad es más antigua de lo que inicialmente se creía; al parecer, culturas ancestrales tenían conocimiento de ella,2 incluso, el mismo Parkinson reconoció que otros autores que le precedieron ya habían descrito signos de la enfermedad.3 Gaceta Médica de México. 2018;154 720 Uno de los datos más antiguos se encuentra en papiros egipcios correspondientes a la Dinastía XIX (1500-1200 a. C.), donde se evoca la sialorrea de los pacientes con esta enfermedad al detallar a un rey al cual “la edad había aflojado su boca” y “escupía continuamente”.4 La Ayurveda (āyuh: ‘duración de la vida’, veda ‘conocimiento’), o antiguo sistema de medicina tradicional de la India, fue publicada alrededor del 1000 a. C. y en ella se menciona a la kampavata (kampa: temblor), enfermedad que se caracteriza por escaso movimiento, exceso de saliva, somnolencia y “mirada reptiliana”.5 Se conoce también que en el Tratado de medicina tradicional china (Nei- Jing), que data de alrededor de 800 a. C., se describen personas con temblor cefálico y de manos, cuya explicación era atribuida de igual forma a la edad.4 En el Antiguo Testamento de la Biblia existen algunas citas interesantes, ejemplo de ello es Eclesiastés 12:3 que dice: “Un día temblarán los guardianes de la casa, y se encorvarán los hombres de batalla; se detendrán las molenderas por ser tan pocas, y se apagarán los que miran a través de las ventanas”, describiéndose la postura y el temblor que podría hacer referencia a EP. Mucho tiempo después, Galeno de Pérgamo, considerado uno de los médicos más influyente de Europa, definió el temblor y pudo distinguirlo entre el de acción y reposo en sus observaciones.5 Lamentablemente, durante siglos desapareció la información relativa a la EP, quizá debido a las numerosas batallas territoriales en las cuales se vio sumergida Europa, que disminuyó la esperanza de vida de los pobladores de esos tiempos, por lo que se podría sospechar que la edad de muerte impedía que la senectud fuera un aspecto común dentro de la sociedad y por ello, probablemente, algunas enfermedades neurodegenerativas pudieron no ser tan frecuentes.6 Las posteriores descripciones que parecen relacionarse con la EP vuelven a emerger a partir del siglo XVII: en 1641, el famoso doctor Nicolaes Tulp retomó el término temblor. Este personaje fue tan importante en su época que le mereció uno de los retratos más conocidos de la historia realizado por el pintor holandés Rembrandt. De igual forma, Silvio de La Boe habló de las características del temblor de acción y en reposo, descripción por la que fue señalado por Parkinson en su ensayo.7 Otras figuras, como Leonardo Da Vinci, Shakespeare o Rembrandt y médicos de la talla de Johanness Baptiste Sagar, Boissier de Sauvages, John Hunter y Chomel hicieron también referencia al temblor y otros trastornos motores en sus obras. En 1758, Gaubius refirió haber observado a “un hombre capaz de correr, pero no de andar”, con lo que aludió a las alteraciones de la marcha que se observan en esta patología.5 Finalmente, en 1817 se publicó el ensayo de James Parkinson, donde por primera vez se recogieron específicamente detalles de la enfermedad, extraídos de la observación de seis pacientes. En ese momento se dio a conocer que el temblor, la bradicinesia y la inestabilidad postural son los signos más importantes de esta entidad. Parkinson indicó que se trataba de un “movimiento tembloroso involuntario… en partes que no están en actividad”, además destacó la “propensión a flexionar el tronco hacia adelante”.3 Al describir la historia natural de la enfermedad, con una redacción claramente coherente y sencilla, reveló que el inicio de la enfermedad era lentamente progresivo, que comenzaba de forma unilateral y que en poco tiempo comprometía el lado contralateral del cuerpo, con posterior compromiso de la posición erecta al caminar y desarrollo de anteropulsión de la marcha. Mencionó que a medida que el tiempo avanzaba, el paciente sentía que los movimientos perdían precisión y los realizaba con gran dificultad. Las caídas se hacían más notorias por la incapacidad de elevar fácilmente las piernas y el lenguaje se volvía inteligible, por lo que el paciente requería la presencia permanente de un cuidador. Quizás una de sus consideraciones con más significación en la actualidad fue la descripción de algunos síntomas no motores de la enfermedad, por ejemplo, las alteraciones del sueño, el estreñimiento, la sialorrea y las alteraciones de los esfínteres. James Parkinson encontró numerosos detractores, aunque finalmente las observaciones directas de afamados médicos de la época resultaron ser útiles para refinar los datos diagnósticos, como sucedió con Jean-Martin Charcot, quien no pudo obtener una versión del referido “Essay on the shaking palsy” traducido al francés sino hasta 1881, gracias a la tarea de traducción que él mismo encomendó a sus estudiantes, luego de lo cual emitió el siguiente comentario: Esta es una definición descriptiva y vívida que es correcta en muchos casos, la mayoría de hecho, y siempre tendrá la ventaja sobre otros de haber sido el primero, pero se equivoca por ser demasiado general. Charcot es quien relacionó por primera vez la rigidez con la enfermedad y señaló que es el signo cardinal de la misma, logrando diferenciarla de la espasticidad.8 De esta manera, parecía que se había Arredondo-Blanco K, et al.: 200 años de Parkinson 721 alcanzado el conocimiento para describir la enfermedad, pero bastaría que los siglos transcurrieran para evidenciar que otras manifestaciones, actualmente denominadas como síntomas no motores, pertenecen a la misma entidad.
Historia de la anatomía e histopatología Después de la descripción clínica de James Parkinson en 1817, así como de los escritos de Jean Martin Charcot titulados “Leçon sur le maladies du systeme nerveux”, uno de los acontecimientos más importantes para la compresión de la EP fue el descubrimiento de la pérdida neuronal en la pars compacta de la sustancia nigra. Esta pérdida fue identificada como la lesión patológica que caracteriza a la EP, lo cual se mantiene hasta la fecha.9 A principios del siglo XX se comenzó a investigar sistemáticamente los mecanismos patogénicos de la enfermedad. En 1913, el patólogo Friederich Lewy describió las inclusiones citoplásmicas de alfa-sinucleína localizadas en neuronas del núcleo motor dorsal del nervio vago y del núcleo basal de Meynert. Estas inclusiones recibieron posteriormente la denominación de cuerpos de Lewy.10 Seis años más tarde, en 1919, Konstantine Trétiakoff dio nombre a la sustancia nigra, detalló su localización y asoció la pérdida de neuronas en esta zona como parte de los hallazgos de la EP.11 En 1960, con el uso de microscopia electrónica, se demostró la estructura interna filamentosa de los cuerpos de Lewy12 y es hasta 2003 cuando Braak et al. publicaron los resultados de los estudios seriados que realizaron en cerebros post mortem, donde evidenciaron la presencia de cuerpos de Lewy en diversas zonas del sistema nervioso, diseñando además un modelo para estadificar la progresión de la enfermedad y su relación con los síntomas motores y no motores.13 En la década de 1980 se reconocieron los efectos neurotóxicos en un grupo de adictos a una “nueva heroína sintética”. Estos sujetos desarrollaron secundariamente un parkinsonismo relacionado con lesión en la sustancia nigra. Fue así como se identificó al MPTP (1-metil-4-fenil-1,2,3,6 tetrahidropiridina) como el agente causal; posteriormente este hallazgo dio origen a los modelos experimentales de EP en animales, que hasta la fecha continúan siendo de gran utilidad.14 En la actualidad se ha descrito no solo la presencia de depósitos de alfa-sinucleína, sino también de proteína tau y de beta amiloide, lo que ha llevado al uso del término de proteinopatías, donde probablemente la patología predominante es la que distinga fenotípicamente a la EP de otras enfermedades como taupatías, Alzheimer o demencia frontotemporal.13 En la última década se ha documentado una transmisión célula a célula de la alfa-sinucleína, al menos en modelos animales y celulares, lo que ha llevado a considerar la posibilidad de que la EP sea un trastorno similar a las enfermedades priónicas.15 Este concepto, de comprobarse, no solo será de gran relevancia para la comprensión de la fisiopatología de la enfermedad, sino también ofrecerá la oportunidad de desarrollar terapia molecular para modificar la progresión de la enfermedad.16 Historia de la genética de la enfermedad de Parkinson Durante décadas se consideró que la EP era una patología de presentación exclusivamente esporádica, ya que aun cuando existían evidencias de un componente familiar, no se disponían de técnicas científicas que permitieran comprobar tal fenómeno. Sin embargo, la evolución tecnológica permitió que este paradigma cambiara. Las primeras investigaciones genéticas sobre esta patología se iniciaron a finales de la década de 1990, en las que se asumió que al menos un porcentaje de los pacientes con EP tiene una causa genética a pesar de la discordancia monocigótica en gemelos que demostraban los estudios de esa época. En 1997, al estudiar cuatro grandes familias con EP se descubrió que una mutación en el gen que codifica la alfa sinucleína ocasiona una forma de herencia dominante de la EP17 y se logró establecer una relación entre los acúmulos de alfa sinucleína y los cuerpos de Lewy.18 Como consecuencia, en los siguientes años esta proteína se convirtió en el blanco de numerosos estudios de investigación. Los parkinsonismos juveniles también se beneficiaron de las técnicas de estudio de material genético. En 1998 se descubrió que mutaciones en la proteína ahora denominada parkina eran la causa de una forma de presentación temprana de la enfermedad.19 En 2003 se logró identificar mutaciones en el gen DJ1 y se asoció como causa de parkinsonismo de inicio temprano.20 PINK 1 fue descubierto en 2004, sugiriéndose como la segunda causa más común de parkinsonismo autosómico recesivo.21 En este mismo año, se reconoció el LRRK2 mediante el mapeo de recombinación de alta resolución y la secuenciación de Gaceta Médica de México. 2018;154 722 genes candidatos en 46 familias, con lo que se iniciaron largos estudios de investigación.22 El crecimiento en el conocimiento genético ha permitido entender la fisiopatología y fenomenología de la EP, además de generar interés en estrategias como terapia génica y reprogramación celular para el manejo de la enfermedad a futuro. A la fecha se conocen 17 genes diferentes considerados como causales, ya sea de forma autosómica dominante o recesiva; adicionalmente se ha identificado un gran número de genes, que, aunque no causales, son considerados de riesgo. Historia del tratamiento de la EP Existen descripciones en la literatura que datan de casi 3000 años atrás, desde la medicina ayurvédica. El tratado clásico médico hindú hace referencia sobre la enfermedad de kampavata, la cual se trataba con un conjunto de remedios vegetales derivados de plantas del género mucuna (Mucuna pruriens), con alto contenido de levodopa obtenida al triturar las semillas. La mucuna es una liana de las selvas tropicales de Asia y América, por lo cual se utilizó también en la región amazónica. En la medicina ayurvédica se daba a los enfermos un brebaje de polvo de semillas de mucuna cocido en leche de vaca, que mejoraba el temblor, la bradicinesia, la rigidez y los calambres, si bien se observaba la presencia de sialorrea. Recientemente se ha demostrado que cada dosis de polvo de semillas contiene alrededor de 200 mg de levodopa.23 En escritos de medicina herbolaria antigua se encuentra información sobre tratamientos contra el temblor con semillas de estramonio, por su efecto anticolinérgico. Las habas y otras legumbres exóticas también contienen levodopa y se utilizan desde hace varios siglos en el tratamiento de la EP.24,25 Por otra parte, desde 1887 Wilhelm Erb introdujo el uso terapéutico de la escopolamina y preparaciones similares fueron utilizadas por sus efectos farmacológicos anticolinérgicos en la década de 1950. Ordestein, discípulo de Charcot, en su tesis médica mencionó el uso de alcaloides de la belladona para controlar el temblor.26 Los tratamientos con efectos anticolinérgicos de Charcot proporcionaron las bases farmacológicas de los modernos medicamentos antiparkinsonianos. La oportunidad histórica de analizar los tratamientos primarios de la enfermedad se encontró con el hallazgo de cartas en la biblioteca personal de este eminente neurólogo. Estas cartas cubren un periodo de 15 meses, de enero de 1863 a marzo 1864.27 William Gowers, contemporáneo de Charcot, utilizaba estrategias terapéuticas similares. Adicionalmente, Gowers describió el efecto negativo del estrés, el cansancio físico y mental, recomendando una vida tranquila;28 para el temblor indicaba hiosciamina, arsénico y cáñamo, así como la combinación de Cannabis y opio.29 En 1910, en un laboratorio londinense, fue sintetizada por primera vez la dopamina por los investigadores George Barger y James Ewens. En el mismo laboratorio, Henry Dale le otorgó el nombre de dopamina. Cabe mencionar que la levodopa, precursor de la dopamina, fue sintetizada químicamente por Casimir Funk en 1911.30 Entre 1950 y 1960, Arvid Carlsson, ganador del premio Nobel de Fisiología y Medicina en 2000, cambiaría el rumbo del tratamiento de la EP con sus trabajos en relación con la dopamina. Al investigar los cambios vasculares generados por la reserpina en conejos observó que el efecto adverso generado por esta era la acinesia. Después de arduos trabajos de investigación, logró revertir este efecto con uso de la D, L DOPA (L-3,4 dihidroxifenilalanina.31 Posteriormente se describió que en el estriado la depleción de la dopamina es la responsable de la presencia de un efecto acinético mediado por reserpina. El austriaco Oleh Hornykiewicz, ganador del premio Wolf de medicina en 1979, fue el responsable de esta notable descripción.32,33 Hornykiewicz fue el primero en proponer el uso de levodopa para el tratamiento de la enfermedad.34 Uno de los experimentos más significativos realizados en la farmacología de la levodopa estuvo a cargo de Kathleen Montagu, con estudios en animales. En noviembre de 1957 descubrió la dopamina intracelular y su distribución en el encéfalo, además, notó que la levodopa incrementa los niveles de catecolaminas en el cerebro. De igual forma, en 1958, Montagu describió que la reserpina provoca depleción de dopamina y que la levodopa restaura sus niveles.35 En ese mismo periodo, Hermann Blaschko, en el Laboratorio de Fisiología de la Universidad de Cambridge, propuso que la levodopa y la dopamina son metabolitos intermedios en las biosíntesis de catecolaminas.36 En esa misma época, Peter Holtz descubrió la enzima dopamina descarboxilasa y documentó que la levodopa es sintetizada a dopamina a través de esta enzima.37 En 1967, George Cotzias evidenció la eficacia y seguridad del uso de levodopa en personas con EP, en Arredondo-Blanco K, et al.: 200 años de Parkinson 723 dosis de 4 y 8 g al día; como consecuencia de este ensayo se introdujo la levodopa en el tratamiento de la EP en conjunto con la suma de los inhibidores de la dopamina dexcarboxilasa, que actualmente es uno de los fármacos más utilizados y con mayor beneficio en los síntomas motores.38 La inclusión de las dosis adecuadas de levodopa fue un logro innovador en el manejo de los síntomas de la enfermedad.39 Sin embargo, poco tiempo después se evidenció que su uso prolongado ocasiona complicaciones motoras,40 lo que obligó a buscar otras alternativas distintas para el tratamiento farmacológico.41 Por otra parte, Kendal B. Corbin en 1949 demostró que el trihexifenidilo, un anticolinérgico, es útil en el control de los síntomas motores de la EP,42 prolongando la utilización de estos medicamentos. Un progreso importante en materia de tratamiento farmacológico fue el desarrollo de los agonistas de la dopamina, los inhibidores de las enzimas monoaminooxidasa B y los inhibidores de la catecol-o-metiltrasferasa. Estos medicamentos fueron inicialmente aprobados para su uso en humanos en las décadas de 1970 y1980. La apomorfina fue el primer agonista de la dopamina sintetizado en el siglo XIX, sin embargo, data desde la era de los mayas, quienes en rituales religiosos empleaban un extracto de la raíz y el tronco de una planta acuática (Nymphaea ampla y Nymphaea caerulea) con propiedades afrodisiacas y alucinógenas, y que posteriormente se descubriría que contiene apomorfina.43,44 En 1869, Matthienssen y Wrigth observaron que al deshidratar la morfina con ácido clorhídrico se obtenía una sustancia, que llamaron apomorfina; en ese entonces comenzó su uso terapéutico, inicialmente como antiemético.45,46 En 1884 se propuso para el tratamiento de pacientes con parkinsonismo,47 pero no fue hasta 1951 que Schwab et al. reportaron que la apomorfina mejoraba dramáticamente, pero en forma temporal, los síntomas que presentaban los pacientes con EP.48 Posteriormente, en 1965, Ernst puntualizó la similitud estructural entre la apomorfina y la dopamina.49 En 1970, Cotzias et al. reevaluaron la utilización de apomorfina y reportaron efectividad antiparkinsoniana, sin embargo, la toxicidad y la necesidad de la administración por vía parenteral limitó su utilidad.50 Tiempo después se intentó el uso de apomorfina por vía oral,51 pero esta práctica fue abandonada por la presencia de hiperazoemia en terapias a largo plazo. A mitad de la década de 1980, Andrew Lees y Gerald Stern demostraron que el uso por vía subcutánea mediante una bomba de infusión mejoraba en 50 % el periodo off en los pacientes con EP.52 Las dificultades técnicas de la vía de administración y los efectos adversos limitaron el uso de la apomorfina, pero a su vez promovió el uso de otros agonistas dopaminérgicos. Inicialmente se utilizaron agonistas dopaminérgicos derivados de la ergotamina (pergolide, bromocriptina, cabergolida, lisuride), los cuales cayeron en desuso debido a sus efectos adversos (náuseas, somnolencia, alucinaciones); el uso prolongado está relacionado con mayor riesgo de fibrosis pleural, pericárdica, retroperitoneal y fibrosis de la válvula cardiaca.53-55 Fue entonces cuando el tratamiento de la EP se enfocó al uso de agonistas dopaminérgicos no ergotamínicos, como el pramipexol y el ropirinol.56 Aun cuando su efecto sintomático es menor que el de la levodopa, su principal indicación surgió por la necesidad de disminuir el riesgo de complicaciones motoras en etapas tardías de la enfermedad; los problemas derivados han sido motivo suficiente para crear opciones farmacológicas que generen una estimulación dopaminérgica continua, la cual disminuye la pulsatilidad en los niveles de estimulación dopaminaérgica. La rotigotina, agonista dopaminérgico de aplicación transdérmica diaria, se comenzó a utilizar en este siglo XXI.57,58 De igual forma se desarrollaron presentaciones de liberación prolongada tanto de ropinirol como de pramipexol.59 Actualmente la infusión subcutánea de apomorfina se considera un tratamiento de avanzada para pacientes con enfermedad complicada.60 A principios de 2010 se describieron los síntomas asociados con la enfermedad, los síntomas no motores, con múltiples tratamientos para su control y así mejorar la calidad de vida de los pacientes. Aún continúa la búsqueda de nuevas alternativas y el perfeccionamiento de las opciones existentes. Otra estrategia terapéutica involucraba el uso de inhibidores de enzimas específicas implicadas en el metabolismo de la dopamina, como la monoaminooxidasa B (MAO-B). La inhibición de esta enzima incrementa la concentración de dopamina endógena y secundariamente reduce los síntomas de la EP. El primer inhibidor de la MAO-B utilizado en la EP fue la selegilina (inicialmente conocida como deprenil) y más recientemente la rasagilina.61 Otro medicamento relevante es la amantadina, aprobada en 1976 como antiviral, pero su uso para la EP se basó en un descubrimiento fortuito como agente antiparkinsoniano. Schwab notó un inesperado beneficio en la severidad del temblor, marcha y bradicinesia en pacientes con Gaceta Médica de México. 2018;154 724 EP y en pacientes con parkinsonismo posencefalítico.62 En la actualidad, el desarrollo de nuevos terapias con efecto neuroprotector se dirige hacia los anticuerpos monoclonales.
Terapia quirúrgica en la enfermedad de Parkinson En 1908, el cirujano Victor Alexander Haden Horsley y el ingeniero Robert Henry Clarke, en Londres, Inglaterra, fueron los pioneros en crear el equipo de estereotaxia,63 lo que sería un parteaguas en la cirugía de EP. Después de 1930, pero antes de la Segunda Guerra Mundial, los avances en neurocirugía se encontraban enfocados en realizar lesiones en las vías corticoespinales en distintos niveles para tratar de corregir el temblor, sin embargo, las secuelas eran catastróficas. Meyer describió los efectos que se producen con lesiones en el caudado, globo pálido y en el núcleo lenticular, demostrando que el temblor y la rigidez secundarias a la EP mejoran con estos procedimientos quirúrgicos, sin embargo, la mortalidad y morbilidad eran altas.64 A la par, Bucy, Case, y Klemme se enfocaron en la cirugía ablativa, con la que se lesionaba la corteza cerebral para el tratamiento del temblor parkinsoniano; la secuela de este tipo de cirugía fue la hemiparesia, por lo cual fue abandonada.65,66 Alrededor de 1939 a 1940, con la presencia de los conflictos bélicos a nivel internacional, la ciencia tuvo un camino intrincado. Las hostilidades entre países terminaron en 1945, pero fue hasta 1947 que se aprobaron las técnicas neuroquirúrgicas y la estereotaxia se comenzó a utilizar en humanos para efectuar procedimientos lesionales principalmente en el tálamo y el globo pálido.67 A comienzo de la década de 1950, Cooper, durante un procedimiento quirúrgico en un paciente con parkinsonismo, lesionó accidentalmente la arteria coroidea anterior, por lo que se vio forzado a ligarla para prevenir la formación de un hematoma; sin embargo, este procedimiento ocasionó el inesperado control del temblor y la rigidez en el sitio contralateral a la lesión generada.68 La talamotomía fue la cirugía más utilizada en la década de 1950 a 1960, técnica empleada por Cooper en Estados Unidos y Hassler en Alemania, porque el tálamo ventrolateral era el sitio ideal para el control de los síntomas motores.69 Posterior a esta fecha, entre 1960 y 1970, con la introducción de la levodopa hubo una reducción acentuada en el empleo de la esterotaxia con fines quirúrgicos enfocados a la EP. A mediados de la década de 1980 renacieron las técnicas neuroquirúrgicas. Lauri Laitinen et al. reconocieron las limitaciones de los tratamientos farmacológicos y retomaron los procedimientos ablativos, particularmente la palidotomía posteroventral.70 En ese mismo periodo, específicamente en 1987, se identificó una función importante del núcleo subtalámico en la EP; múltiples equipos de científicos en distintas partes del mundo realizaron trabajos experimentales, los cuales fueron inicialmente realizados en modelos animales con parkinsonismo y posteriormente en humanos afectados con la enfermedad.71-73 Las observaciones de Bergman, Witchman y De Long, en relación con la respuesta con una lesión en el núcleo subtalámico, provee un arma importante para el desarrollo del que tal vez sea considerado uno de los más grandes descubrimientos de esa época: la estimulación cerebral profunda de alta frecuencia.74 El año de 1993 se considera un parteaguas en relación con la cirugía funcional: Alim-Louis Benabid reportó la implantación de electrodos en el núcleo subtalámico.75 Por lo anterior, se considera a 1987 como el año del nacimiento de la estimulación cerebral profunda y a 30 años de esta valiosa aportación, se dispone de diversos procedimientos de neurocirugía funcional y de estimulación cerebral profunda, los cuales han evolucionado a la par de las tecnologías contemporáneas, que también progresan incesantemente.
Aún queda mucho camino por recorrer, y es responsabilidad de los científicos y médicos de la actualidad continuar escribiendo la historia día a día con el fin de encontrar la cura, que nos ha eludido durante los últimos dos siglos.
Bibliografía
Keppel-Hesselink JM. Evolution of concepts and definitions of Parkinson’s disease since 1817. J Hist Neurosci. 1996; 5:200-207. 2. Hurwitz B. Urban observation and sentiment in James Parkinson’s essay on the shaking palsy (1817). Lit Med. 2014;32:74-104. 3. Parkinson J. Definition, history, illustrative cases. En: An essay on shaking palsy. Londres: Whittingham and Rowland; 1817. 4. León R. Historia de la levodopa; un tratamiento anunciado desde la antigüedad. AFT. 2007;5:205-207. 5. García P. Prehistoria de la enfermedad de Parkinson. Neurologia. 2004;19:735-737. 6. Jonker MA. Estimation of life expectancy in the Middle Ages. J R Statist Soc A. 2003;166:105-117. 7. Rosler R, Young P. La lección de anatomía del doctor Nicolaes Tulp: el comienzo de una utopía médica. Rev Med Chile. 2011;139:535-541. 8. Charcot JM. Lecture VII: On slow compression of the spinal cord. En: Lectures on the diseases of the nervous system. Londres: New Sydenham Society; 1881. 9. Charcot JM. De la paralysie agitante. En: Oeuvres complètes. Leçons sur les maladies du système nerveux. Tomo I. París: Bureaux du Progrès Médical; 1892. pp. 155-188. 10. Lewy FH. Zur pathologischen Anatomie der Paralysis agitans. Deutsche Zeitschrift für Nervenheilkunde. 1913;50:50-55. 11. Trétiakoff K. Contribution à l’étude de l’anatomie pathologique du locus niger de Soemmering avec quelques déductions relatives à la pathogénie des troubles du tonus musculaire et de la maladie de Parkinson. Francia: Universidad de París; 1919. 12. Bethlem J, Den-Hartog-Jager WA. The incidence and characteristics of Lewy bodies in idiopathic paralysis agitans (Parkinson’s disease). J Neurol Neurosurg Psychiatry. 1960;23:74-80. 13. Braak H, Del-Tredici K, Rüb U, De-Vos RA, Jansen-Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197-211. 14. Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979-980. 15. Angot E, Steiner JA, Hansen C, Li JY, Brundin P. Are synucleinopathies prion-like disorders? Lancet Neurol. 2010;9:1128-1138. 16. Hasegawa M, Nonaka T, Masuda-Suzukake M. Prion-like mechanisms and potential therapeutic targets in neurodegenerative disorders. Pharmacol Ther. 2017;172:22-33. 17. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dutra A, Pike B, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045-2047. 18. Nussbaum RL, Polymeropoulos MH. Genetics of Parkinson’s disease. Hum Mol Genet. 1997;6:1687-1691. 19. Jones AC, Yamamura Y, Almasy L, Bohlega S, Elibol B, Hubble J, et al. Autosomal recessive juvenile parkinsonism maps to 6q25.2-q27 in four ethnic groups: detailed genetic mapping of the linked region. Am J Hum Genet. 1998;63:80-87. 20. Bonifati V, Rizzu P, Van-Baren MJ, Schaap O, Breedveld GJ, Krieger E, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256-259. 21. Hatano Y, Li Y, Sato K, Asakawa S, Yamamura Y, Tomiyama H, et al. Novel PINK1 mutations in early-onset parkinsonism. Ann Neurol. 2004;56:424-427. 22. Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601-607. 23. Katzenschlager R, Evans A, Manson A, Patsalos PN, Ratnaraj N, Watt H, et al. Mucuna pruriens in Parkinson’s disease: a double blind clinical and pharmacological study. J Neurol Neurosurg Psychiatry. 2004; 75:1672-1677. 24. Guebila MB, Thiele I. Model-based dietary optimization for late-stage, levodopa-treated, Parkinson’s disease patients. NPJ Syst Biol Appl. 2016;2:16013. 25. Nagashima Y, Kondo T, Sakata M, Koh J, Ito H. Effects of soybean ingestion on pharmacokinetics of levodopa and motor symptoms of Parkinson’s disease: in relation to the effects of Mucuna pruriens. J Neurol Sci. 2016;361:229-234. 26. Ordenstein L. Étiologie, pronostic, thérapeutique. En: Sur la paralysie agitante et la sclérose plaque généralisée. Francia: A Delahaye, Libraire-Éditeur; 1868. 27. Charcot JM. Leçon 5. En: Oeuvres complètes de J.M. Leçons sur les maladies du système nerveux. Tomo I. Bureaux du Progrès Médical; París: 1892. 28. Gowers WR. Paralysis agitans. En: A system of medicine. Londres: Macmillan; 1899. 29. Gowers WR. Paralysis agitans. En: A manual of diseases of the nervous system. Londres: J. & A.; 1888. 30. Fahn S. The history of dopamine and levodopa in the treatment of Parkinson’s disease. Mov Disord. 2008;23:497-508. 31. Carlsson A, Waldeck B. A fluorimetric method for the determination of dopamine (3‐hydroxytyramine). Acta Physiol Scand. 1958;44:293-298. 32. Price KS, Farley IJ, Hornykiewicz O. Neurochemistry of Parkinson’s disease: relation between striatal and limbic dopamine. Adv Biochem Psychopharmacol. 1978;19:293-300. 33. Lloyd K, Davidson L, Hornykiewicz O. The neurochemistry of Parkinson’s disease: effect of L-dopa therapy. J Pharmacol Exp Ther. 1975;195:453-464. 34. Ehringer H, Hornykiewicz, O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system. Parkinsonism Relat Disord. 1960;4:53-57. 35. Montagu KA. Catechol compounds in rat tissues and in brains of different animals. Nature. 1957;180:244-245. 36. Blaschko H. The specific action of L-dopa decarboxylase. J Physiol. 1939;96:50-51. 37. Holtz P. Dopa decarboxylase. Naturwissenschaften. 1939;27:724-725. 38. Cotzias GC, Van-Woert MH, Schiffer LM. Aromatic amino acids and modification of parkinsonism. N Engl J Med. 1967;276:374-379. 39. Cotzias GC, Papavasiliou PS, Gellene R. Modification of Parkinsonism: chronic treatment with L-dopa. N Engl J Med. 1969;280:337-345. 40. Fahn S. Parkinson disease the effect of levodopa and the ELLDOPA trial. Earlier vs later L-dopa. Arch Neurol. 1999;56:529-535. 41. Doshay LJ, Constable K. Newer drugs in the treatment of parkinsonism. Neurology. 2001;57:4-10. 42. Corbin KB. Trihexyphenidyl; evaluation of the new agent in the treatment of Parkinsonism. J Am Med Assoc. 1949;141:377-382. 43. Bertol E, Fineschi V, Karch SB, Mari F, Riezzo I. Nymphaea cults in ancient Egypt and the New World: a lesson in empirical pharmacology. J R Soc Med. 2004;97:84-85. 44. Taba P, Lees A, Stern G. Erich Harnack (1852-1915) and short history of apomorphine. Eur Neurol. 2013;69:321-324. 45. Matthiessen A. Researches into the chemical constitution of the opium bases. Part I.- On the action of hydrochloric acid on morphia. Proc R Soc Lond. 1868;17:455-460. 46. Matthiessen A, Wright C. Apomorphia, a new base derived from morphine. Pharm J Tr. 1869;11-12. 47. Djamshidian A, Poewe W. Apomorphine and levodopa in Parkinson’s disease: two revolutionary drugs from the 1950’s. Parkinsonism Relat Disord. 2016;33:S9-S12. 48. Schwab RS, Amador LV, Lettvin JY. Apomorphine in Parkinson’s disease. Trans Am Neurol Assoc. 1951;56:251-253. 49. Ernst AM. Relation between the action of dopamine and apomorphine and their O-methylated derivatives upon the CNS. Psychopharmacologia. 1965;7:391-399. 50. Cotzias GC, Düby S, Ginos JZ, Steck A, Papavasiliou PS. Dopamine analogues for studies of parkinsonism. N Engl J Med. 1970;283:1289. 51. Cotzias GC, Papavasiliou PS, Tolosa ES, Mendez JS, Bell-Midura M. Treatment of Parkinson’s disease with aporphines: Possible role of growth hormone. N Engl J Med. 1976;294:567-572. 52. Stibe C, Lees A, Stern G. Subcutaneous infusion of apomorphine and lisuride in the treatment of parkinsonian on-off fluctuations. Lancet. 1987;1:871. 53. Lees AJ, Haddad S, Shaw KM, Kohout LJ, Stern GM. Bromocriptine in parkinsonism: a long-term study. Arch Neurol. 1978;35:503-505. 54. Pearce RK, Banerji T, Jenner P, Marsden CD. De novo administration of ropinirole and bromocriptine induced less dyskinesia than L‐dopa in the MPTP‐treated marmoset. Mov Disord. 1998;13:234-241. 55. Rascol O, Pathak A, Bagheri H, Montastruc JL. Dopaminagonists and fibrotic valvular heart disease: further considerations. Mov Disord. 2004;19:1524-1425. 56. Hobson DE, Pourcher E, Martin WR. Ropinirole and pramipexole, the new agonists. Can J Neurol Sci. 1999;26:S27-S33. 57. Metman LV, Gillespie M, Farmer C, Bibbiani F, Konitsiotis S, Morris M, et al. Continuous transdermal dopaminergic stimulation in advanced Parkinson’s disease. Clin Neuropharmacol. 2001;24:163-169. 58. Pfeiffer RF. Potential of transdermal drug delivery in Parkinson’s disease. Drugs Aging. 2002;19:561-570. 59. Jenner P, Könen-Bergmann, M, Schepers C, Haertter S. Pharmacokinetics of a once-daily extended-release formulation of pramipexole in healthy male volunteers: three studies. Clin Ther. 2009;31:2698-2711. 60. Auffret M, Le-Jeune F, Maurus A, Drapier S, Houvenaghel JF, Robert GH, et al. Apomorphine pump in advanced Parkinson’s disease: Effects on motor and nonmotor symptoms with brain metabolism correlations. J Neurol Sci. 2017;372:279-287. 61. Bar Am O, Amit T, Youdim MB. Contrasting neuroprotective and neurotoxic actions of respective metabolites of anti-Parkinson drugs rasagiline and selegiline. Neurosci Lett. 2004;355:169-172. 62. Schwab RS, England AC, Poskanzer DC, Young RR. Amantadine in the treatment of Parkinson’s disease. JAMA. 1969;208:1168-1170. 63. Foerster O, Penfield W. The structural basis of traumatic epilepsy and results of radical operation. Brain 1930; 53:99-119. 64. Meyers RA. Surgical procedure for the alleviation of postencephalitic tremor, with notes on the physiology of premotor fibers. Arch Neurol Psychiatry. 1940;44:455-459. Gaceta Médica de México. 2018;154 726 65. Bucy PC, Case TJ. Tremor, physiologic mechanism and abolition by surgical means. Archi Neurol. 1939;41:721-746. 66. Klemme RM. Surgical treatment of dystonia, paralysis agitans and athetosis. Arch Neurol Psychiatry. 1940;44:926. 67. Spiegel EA, Wycis HT, Marks M, Lee AJ. Stereotaxic apparatus for operations on the human brain. Science. 1947;106:349-350. 68. Cooper IS. Ligation of the anterior choroidal artery for involuntary movements; parkinsonism. Psychiatr Q. 1953;27:317-319. 69. Hassler R. The influence of stimulations and coagulations in the human thalamus on the tremor at rest and its physiologic mechanism. Proc Second Intl Congr Neuropath. 1:637-642. 70. Laitinen LV, Bergenheim AT, Hariz MI. Leksell’s posteroventral pallidotomy in the treatment of Parkinson’s disease. J Neurosurg. 1992;76:53-61. 71. Smith Y, Parent A. Neurons of the subthalamic nucleus in primates display glutamate but not GABA immunoreactivity. Brain Res. 1988;453:353-356. 72. Rodríguez-Oroz MC, Rodríguez M, Guridi J, Mewes K, Chockkman V, Vitek J, et al. The subthalamic nucleus in Parkinson’s disease: somatotopic organization and physiological characteristics. Brain. 2001; 124:1777-1790. 73. Aziz TZ, Peggs D, Sambrook MA, Crossman AR. Lesion of the subthalamic nucleus for the alleviation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism in the primate. Mov Disord. 1991;6:288-289. 74. Bergman H, Wichmann T, DeLong MR. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249: 1436-1438. 75. Pollak P, Benabid AL, Gross C, Gao DM, Laurent A, Benazzouz A, et al. Effects of the stimulation of the subthalamic n
Kenia Arredondo-Blanco,1 Rosalía Zerón-Martínez,1 Mayela Rodríguez-Violante1 y Amin Cervantes-Arriaga2 1 Clínica de Trastornos del Movimiento; 2 Laboratorio Clínico de Enfermedades Neurodegenerativas. Secretaría de Salud, Instituto Nacional de Neurología y Neurocirugía, Ciudad de México, México Gac Med Mex. 2018;154:719
Los desordenes extrapiramidales y como prototipo la Enfermedad de Parkinson, son un problema creciente,
No hay una terapia eficaz en la enfermedad de Parkinson ni medicamentos que lo puedan evitar el aumento de las enfermedades virales están induciendo un crecimiento de esta enfermedad .
La aparición de algunos casos de Parkisonismo en la Pandemia del Covid 19, nos están asustando.
La bibliografía nos muestra que una serie de procesos microbianos en la historia desencadenaban trastornos extrapiramidales.
El manejo farmacológico de la enfermedad de Parkinson requiere un equilibrio entre optimizar la eficacia clínica y minimizar los eventos adversos3
Un resumen de los medicamentos utilizados en esta patología:
Levodopa es el pilar del tratamiento actual para la enfermedad de Parkinson1,3
Inhibidores de la decarboxilasa de la dopamina (DDC) se administran con levodopa para inhibir su metabolismo periférico1
Los inhibidores de la monoaminooxidasa B (MAOB) a veces se utilizan para el tratamiento inicial de la enfermedad de Parkinson para retrasar la necesidad de terapia con levodopa1,4 y también se puede utilizar en combinación con medicamentos a base de levodopa1
Los agonistas de la dopamina (DA) se pueden utilizar como monoterapia para el tratamiento sintomático inicial en la enfermedad de Parkinson o como complemento de la levodopa1,4
Los inhibidores de la catecol-O-metiltransferasa (COMT) previenen el metabolismo de la levodopa periférica, para aumentar la vida media de la levodopa y su entrega al cerebro5
Los anticolinérgicos se utilizan principalmente para aliviar los síntomas del movimiento leve, particularmente los temblores y la rigidez muscular, en pacientes más jóvenes en las primeras etapas de la enfermedad de Parkinson1
Los antagonistas de los receptores de N-metil-D-aspartato (NMDA) se utilizan clásicamente para reducir la discinesia y controlar los síntomas motores6
Pese a estos tratamientos, los movimientos anormales y su cortejo, son muy dificiples de controlar
Referencias:
Zahoor I, Shafi A, Haq E. Tratamiento farmacológico de la enfermedad de Parkinson. Enfermedad de Parkinson: Patogénesis y Aspectos Clínicos [Internet], eds Stoker TB, Groenlandia JC. Brisbane, Australia: Codon Publications, 2018:129–44.
DeMaagd G, Philip A. Parte 2: introducción a la farmacoterapia de la enfermedad de Parkinson, con un enfoque en el uso de agentes dopaminérgicos. P T 2015;40:590–600.
. Lancet 2014;384:1196–205.
Mejora de la terapia con L-dopa: el desarrollo de inhibidores enzimáticos. Mov Disord 2015;30:103–13.
BORRAR LAS DRUSAS MACULARES PARA PREVENIR LA DMAE
Las drusas maculares aparecen entre la membrana basal del epitelio pigmentario de la retina (EPG) y la membrana de Bruch.
Se presentan como nódulos de color blanco o amarillo, de diferentes tamaños y, si no se asocian a una lesión de la mácula, no suelen producir síntomas. Son los residuos en definitiva que el cuerpo no es capaz de eliminar a través de la circulación sanguínea y se almacenan pudiendo aparecer en el nervio óptico o en la mácula del ojo (zona central de la retina que nos permite percibir los detalles).28 ene. 2018
Las inyecciones de líquido subretiniano son una terapia emergente para eliminar drusas maculares y prevenir la degeneración macular asociada a la edad (DMAE).
Hasta fechas recientes poco se podía hacer para eliminar las drusas, que en muchos casos son un paso inicial en la degeneración macular asociada a la edad (DMAE), una enfermedad que altera la visión y que, en sus formas más graves, progresa a ceguera. “Las drusas mayores de 125 micras tienen riesgos”, advierte Hugo Quiróz-Mercado, exdirector del Laboratorio de Cirugía Experimental del Hospital Luis Sánchez de la Asociación para la Prevención de la Ceguera (México). Este oftalmólogo relata que las drusas pueden verse incluso en pacientes con agudeza visual 20/20 y ya operados de cataratas.
“A los pacientes podemos decirles que no fumen, que coman zanahorias, que cuiden su salud, pero su visión irá deteriorándose hasta desembocar en una degeneración macular húmeda, que es la que ocasiona la grave y rápida disminución de la agudeza visual”. De hecho, supone en torno al 10 por ciento de todas las DMAE, pero en el 90 por ciento de los casos conduce a ceguera legal.
“Los depósitos que se forman bajo el epitelio pigmentario retiniano (EPR) tienen una fisiopatología muy compleja desde el punto de vista bioquímico y genético, pero siempre me ha interesado la posibilidad de hacer algo por estos pacientes bioquímica o quirúrgicamente”, ha explicado durante el congreso FacoElche 20/20 al exponer las alternativas disponibles -muchas de ellas experimentales- para poder tratar estas drusas. Grosso modo, las drusas son unos depósitos amarillos que se forman entre el EPR y la membrana de Bruch, y que dependiendo de su tipología producen manchas borrosas o negras en el campo visual.
A día de hoy se ensayan diferentes terapias a base de inyecciones subretinianas, energía láser subumbral, vitrectomía pars plana y medicamentos para la DMAE húmeda. “No es ninguna locura introducir líquido subretiniano en los agujeros maculares, pues se ha visto que da elasticidad a la retina”, avanza el experto, que confía menos en el láser subumbral porque, a pesar de disminuir las drusas los pacientes siguen desembocando en una DMAE húmeda o seca. No obstante cree que separando los pacientes con drusas de los que tienen depósitos drusoides, podría tratarse con más precisión. La tomografía de coherencia óptica (OCT) revela que el 14 por ciento de los pacientes con drusas tienen oculta una membrana neovascular coroidea –vasos sanguíneos nuevos y dañinos que crecen en la coroides-. Eso podría ser el impedimento para prevenir la DMAE con láser subumbral.
También quiso repasar lo que ocurre con otras cirugías que logran eliminar drusas, aunque con casos individualizados. Así, un paciente con agudeza visual (AV) de 20/60 y drusas al que se le desprende la retina y presenta una AV de 20/100, pero al año de la operación su AV es de 20/25 y las drusas han disminuido.
“Estamos viendo que en pacientes con agujero macular las drusas se modifican, o sea, que algo pasa con la cirugía”, incide el oftalmólogo, recordando el estudio de otro paciente a quien le desaparecieron las drusas en el ojo intervenido por agujero macular (pasó de AV 20/200 a 20/25), pero en el ojo no operado siguió con 20/100. “Y con el síndrome de tracción vítreomacular pasa algo muy interesante. Al 60 por ciento de los pacientes que además tienen drusas les desaparecen simplemente haciéndoles la vitrectomía. La explicación podría ser que al quitar el vítreo se oxigena la retina.
Uno de los objetivos futuros es saber qué sucede en el espacio subretiniano a nivel molecular
“Aún no sabemos realmente qué estímulos creamos en el EPR para que se regenere y desaparezcan las drusas” . Quiróz-Mercado se pregunta qué sucede en el espacio subretiniano a niveles moleculares. Provocando desprendimiento de retina en ratones se elevan las fibulinas, pequeños péptidos relacionados con la presencia de drusas. Y en una investigación del Hospital de la Ceguera demuestran que las fibulinas están estrechamente relacionadas con las anti-integrinas y otros péptidos de la matriz intracelular. “Estamos a punto de desembocar en un tratamiento experimental sobre la presencia de drusas”, confía el experto.
En otros campos de investigación ya se ha visto que las fibulinas inhiben el cáncer de mama y de colon, así como la angiogénesis característica de cáncer y procesos neurodegenerativos
Otros tratamientos singulares que alcanzan resultados pioneros son la inyección subretinal de solución salina balanceada para irrigación oftálmica, que inicialmente muestra beneficios superiores a la terapia génica; o, en algunos casos, si no desaparecen las drusas con la inyección, los pacientes podrían beneficiarse de cirugía, con vitrectomía. “Sería interesante que pudiéramos inyectar una sustancia con péptidos mediadores que propicien un mejor funcionamiento del EPR”, sugiere el experto.
Aparecen aquí nuevas pistas. El uso de terapias anti-integrinas en diabetes se relaciona con el estrés oxidativo, pero también con la neurodegeneración en retinopatía diabética. En algunos trabajos se ha visto menos isquemia cuando se usa terapia anti-integrinas en modelos animales. En el laboratorio experimental de Quiróz-Mercado utilizaron células neuronales y de EPR y se comprobó una sobrevida celular si se someten a estrés con un ácido determinado, incluso con peróxido. Secuencialmente otros investigadores han visto con el mismo tratamiento una protección de las mitocondrias, o un efecto neuroprotector de las anti-integrinas. Esto último también se ha experimentado cultivando células de epitelio pigmentario humano, incluso se ha visto en expresión genética.
Ellos han provocado daño en el nervio óptico de ratas y las han tratado con y sin integrinas, resultando que las células ganglionales están más preservadas en el grupo de las anti-integrinas. En 2017, su equipo de investigación logró mejorar la visión con una terapia anti-integrinas en 12 pacientes que presentaban drusas y disminución de la agudeza visual.
Un estudio multicéntrico de la Universidad de Miami concluye que todos los pacientes que recibieron inyección intravítrea de anti-integrinas mejoraron la agudeza visual.










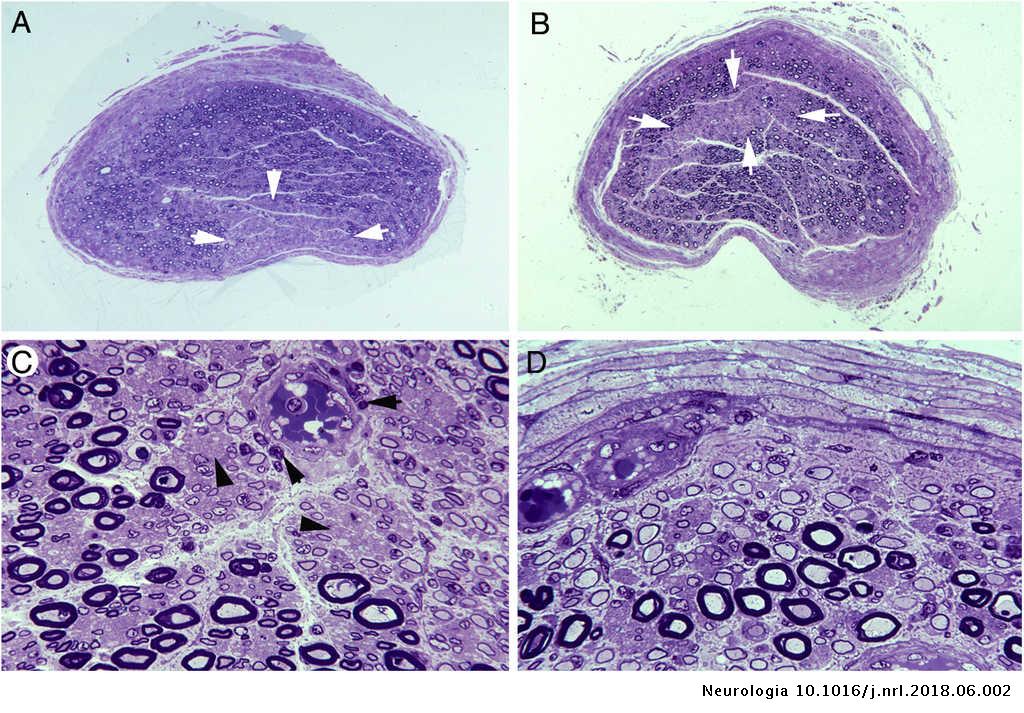
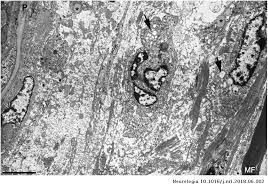



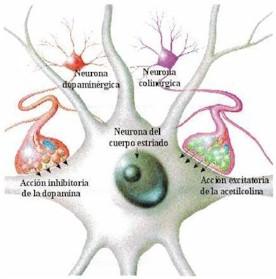

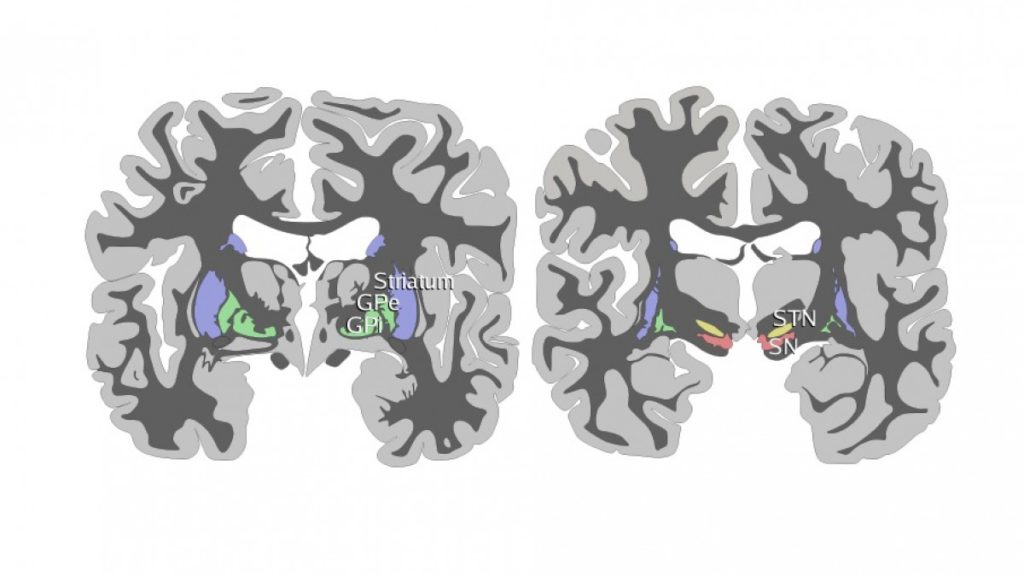
 BORRAR LAS DRUSAS MACULARES PARA PREVENIR LA DMAE
BORRAR LAS DRUSAS MACULARES PARA PREVENIR LA DMAE