UN MECANISMO TOXICO CAUSA LA ELA


.Un nuevo mecanismo tóxico que bloquea cualquier reacción celular que use ácidos nucleicos originaría la muerte de las neuronas motoras en pacientes con ELA.
La esclerosis lateral amiotrófica (ELA), la “enfermedad de Lou Gehrig”, es una enfermedad neurodegenerativa progresiva que afecta a las células nerviosas del cerebro y de la médula espinal. Las neuronas motoras van del cerebro a la médula espinal y de la médula espinal a los músculos de todo el cuerpo.
A-mio-trófica proviene del griego. «A» significa sin o carente. «Mio» se refiere a los músculos, y «trófica» significa alimentación: «Sin alimentación a los músculos». Cuando un músculo no es alimentado, se «atrofia» o se desgasta. «Lateral» identifica las áreas de la médula espinal donde se localizan partes de las células nerviosas que dan señales y controlan los músculos. A medida que esta área se va degenerando, produce la cicatrización o el endurecimiento («esclerosis») en la región.
A medida que las neuronas motoras se van degenerando, dejan de enviar impulsos a las fibras musculares que normalmente resultan en el movimiento muscular. Los primeros síntomas de la ELA a menudo incluyen una mayor debilidad muscular, especialmente en brazos y piernas, en el habla, en la acción de tragar o en la respiración. Cuando los músculos dejan de recibir los mensajes de las neuronas motoras que requieren para funcionar, se empiezan a atrofiar (se vuelven más pequeños). Las extremidades se empiezan a ver más «delgadas», a medida que se atrofia el tejido muscular.
Astas anteriores de la médula espinal con atrofia, degeneración y cromatolisis de neuronas
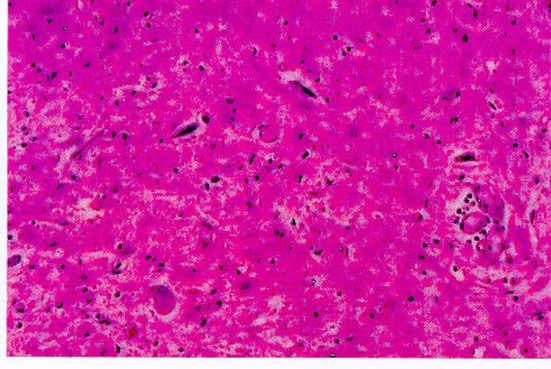
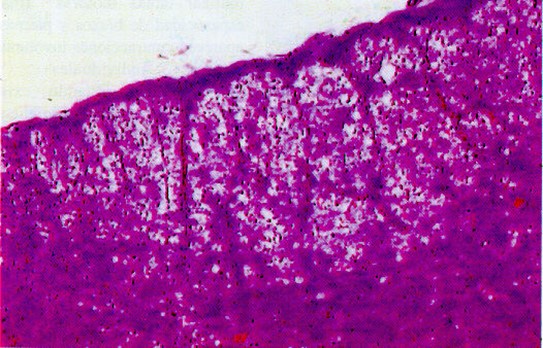
Desmielinización de cordones laterales de medula espinal
FACTORES de riesgo EN LA esclerosis lateral amiotrófica:
Factor hereditario. Entre el 5 y el 10 % de las personas con esclerosis lateral amiotrófica la heredaron (esclerosis lateral amiotrófica familiar). La mayoría de los hijos de personas con esclerosis lateral amiotrófica familiar tienen un 50 % de probabilidades de desarrollar la enfermedad.
La edad. El riesgo de esclerosis lateral amiotrófica aumenta con la edad, y es más común entre los 40 y mediados de los 60 años.
Sexo. Antes de los 65 años, la esclerosis lateral amiotrófica es un poco más común en hombres que en mujeres. Esta diferencia de sexo desaparece después de los 70 años.
La genética. Algunos estudios que examinan todo el genoma humano encontraron muchas similitudes en las variaciones genéticas de las personas con esclerosis lateral amiotrófica familiar y algunas personas con esclerosis lateral amiotrófica no hereditaria. Estas variaciones genéticas podrían hacer que las personas sean más susceptibles a la esclerosis lateral amiotrófica.
Se trata de la tercera enfermedad neurodegenerativa con mayor incidencia –después de la demencia y el Parkinson-, y de elevada mortalidad. Se calcula que cada año aparecen unos 2-5 casos por cada 100.000 habitantes en el mundo. En España, se producen tres nuevos pacientes diagnosticados cada día.
Actualmente, carece de tratamiento y su causa es aún una incógnita. No obstante, se sabe que en el 10% de los afectados hay un importante componente genético, que hace que la enfermedad aparezca en varios miembros de una familia.
En casi la mitad de estos casos de ELA familiar el origen está en un gen llamado C9ORF72 en el cual, sus mutaciones tienen un papel destacable.
La pregunta clave es por qué las mutaciones en este gen destruyen a las neuronas motoras. Según Óscar Fernández-Capetillo, responsable del Grupo de Inestabilidad Genómica del Centro Nacional de Investigaciones Oncológicas (CNIO), en Madrid, se ha descubierto un mecanismo que explica la toxicidad derivada de mutaciones en el gen C9ORF72.
“A principios de 2011 se descubrió que la mutación más frecuente en pacientes de ELA se encuentra en este gen. Pocos años después, se confirma que esta mutación acaba produciendo unos pequeños péptidos que son tóxicos, pero se desconocía el mecanismo que conducía a ello y que ahora se ha identificado en el CNIO»,
En la mutación del C9ORF72 de los pacientes con ELA existe una pequeña secuencia repetida de ADN que, en personas no enfermas suele ser de unas 8-10 copias, y que en estos pacientes está expandida hasta ciento de veces. “Esta expansión de repeticiones se traducen en proteínas que generan unos pequeños péptidos que son tóxicos, un fenómeno que, por otra parte, se ha observado en otros contextos de la naturaleza humana y para el que hemos desarrollado un modelo que los conecta a todos y explica así estos problemas tan generalizados”.
El mecanismo tóxico identificado y que está asociado a un gen concreto, el C9ORF72, “no se descarta la posibilidad que otras mutaciones relacionadas con ELA estén actuando de manera similar, es decir, bloqueando el ADN y ARN de las neuronas motoras”.
Aprender a aliviar la toxicidad de estos péptidos puede ser útil también para abordar los casos de ELA no asociados a C9ORF72, lo que englobaría a la enfermedad en su conjunto.
“la gran mayoría de las mutaciones halladas en pacientes de ELA son en proteínas que se unen a ARN, y generalmente lo que hacen estas mutaciones es impedir la unión de estas proteínas al ARN».
Además, las células de estos pacientes también tienen problemas muy generales con sus ácidos nucleicos, por lo que «pensamos que, aunque las mutaciones en C9ORF72 afectan a solo una parte de los pacientes de ELA, el mecanismo subyacente a la toxicidad de las neuronas puede que no sea muy diferente, en lo fundamental, a lo que pasa en el resto de los casos de esta enfermedad”.
“
Bibliografía
Crawford TO. From enigmatic to problematic. The new molecular genetics of childhood spinal muscular atrophy. Neurology 46: 335-340, 1996.
Ikeda M et al: variable clinical symptoms in familiar amyotrofic lateral sclerosis with a novel point in the Cu/Zn superoxide dismutase gene. Neurology 45: 2038-2041, 1995.
Rosen DR et al: Mutations in Cu/Zn superoxide dismutase gene are associated with familiar amyotrofic lateral sclerosis. Nature 362: 59, 1993.
Siddique T et al: Linkage analysis in familial amyotrofic lateral sclerosis, Neurology 39: 919-926, 1989.
Veltema AN, Ross RAC, and Bruyn GW: Autosomal dominant adult amyotrofic lateral sclerosis, J Neurol Sci 97: 93-115, 1990.
LaSpada AR, et al: Androgen receptor gene mutations in X linked spinal and bulbar muscular atrophy, Nature 352: 77-79, 1991.
Rohstein JD, et al: Abnormal excitatory amino acid metabolism in amyotrofic lateral sclerosis, Ann Neurol 28: 18-25, 1990.
Younger DS, et al: Motor neuron disease and ALS: Relation of high CSF protein content to paraproteinemia and clinical syndromes, Neurology 40: 595-599, 1990.
Williams C, et al: Degeneration of spinocerebellar neurons in amyotrofic lateral sclerosis, Ann Neurol 27: 215-225, 1990.
Brownell B, Oppenheimer DR, and Hughes TJ: The central nervous system in motor neuron disease, J Neurol Neurosurgery Psychiatry 33: 338-357, 1970.
Alvarado F Rodriguez V Esclerosis Lateral amiotrófica. Casos familiares en 6 hermanos. En preparación. Archivos del Departamento de Patología. Hospital México.
Cotran RS, Kumar V, Collins T. Robbins. Patologia estructural y funcional. 6ª ed. Madrid.- McGraw-Hill/Interamericana, 2000.
Neuropathology The diagnostic Approach. St Louis: Mosby, 1977. Raquel Serrano
Vanesa Lafarga,
.