ARTERIOSCLEROSIS Y GERMENES
La arterioesclerosis es un proceso en el que depósitos de material graso llamado placa se acumulan en las paredes de las arterias reduciendo o bloqueando el flujo sanguíneo 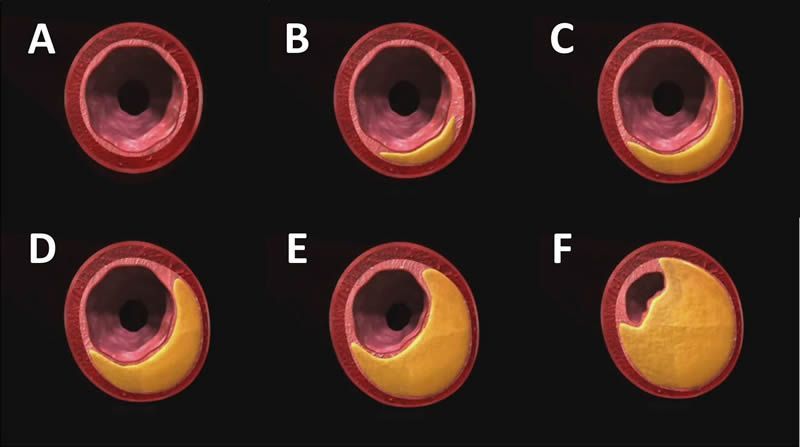
De A hacia F, formación y progresión de la placa de ateroma. En las fases iniciales (A-C) hay aumento del tamaño de la arteria por remodelado con escasa afectación de la luz y en las fases finales (D-F) se produce la estenosis de la arteria.
| Los factores de riesgo clásicos de la arteriosclerosis son el tabaquismo, la diabetes, la hipertensión y los niveles elevados de colesterol, y que los factores genéticos también intervienen en el origen de la enfermedad. Sin embargo, también señalan que en algunos casos tienen lugar episodios cardiovasculares sin la presencia de los citados factores de riesgo. Desde hace un tiempo, se citan la inflamación y los agentes infecciosos como factores que también tienen un papel en el inicio del proceso aterosclerótico.
La etiología infecciosa de la arteriosclerosis hace mucho tiempo que preocupa y son varios los gérmenes que se han involucrado en su desarrollo.
No sería atrevido incluir la arterioesclerosis en las enfermedades degenerativa, donde una noxa extraña pone en marcha un sistema de reparación inapropiado.
|
La respuesta inflamatoria está mediada por las sustancias producidas por los macrófagos y linfocitos T. Estas sustancias (factor de crecimiento derivado de las plaquetas, factor de crecimiento de los fibroblastos, etc.) provocan una migración de células musculares lisas y su posterior proliferación. Unas células musculares lisas conservan su función contráctil y otras la pierden y son capaces de producir matriz extracelular, constituyendo la capa fibrosa que envuelve al núcleo lipídico. Los monocitos al atravesar la pared arterial se diferencian en macrófagos y sintetizan una gran cantidad de sustancias que se van a encargar de perpetuar la respuesta inflamatoria y estimular el sistema inmune. Una de sus funciones principales es fagocitar a las lipoproteínas de baja densidad (LDL), acción mediada por las LDL oxidadas (LDL-ox), la interleucina 1 y el factor de necrosis tumoral alfa (TNF-*), entre otras, constituyendo las células espumosas que son el sustrato anatómico del núcleo lipídico de la placa de ateroma. Los linfocitos T son atraídos por la acción del TNF-, la interleucina 2 y el factor estimulante de colonias de los granulocitos/macrófagos. Éstos son los grandes desconocidos de todo el proceso, y actualmente una de las múltiples líneas de investigación se centra en intentar esclarecer su papel en el proceso aterosclerótico. Se sabe que los linfocitos T cooperadores (CD4) predominan en las placas maduras y que los linfocitos T supresores (CD8) predominan en las lesiones precursoras de la placa o en la periferia de las lesiones9.
Esto condiciona la adhesión y formación de un trombo que, al ocluir la luz de un vaso , va a provocar la isquemia distal a la lesión. La ruptura de la placa suele producirse por la acción de metaloproteasas, como las colagenasas producidas por los macrófagos activados de la placa que van adelgazando la capa fibrosa8
Los factores de riesgo cardiovascular clásicos (hipertensión arterial, diabetes mellitus, hipercolesterolemia, tabaco, homocisteína, predisposición genética) y otros más recientes, como determinadas infecciones, provocan una disfunción endotelial que desencadenará el fenómeno aterosclerótico. A su vez la célula endotelial es capaz de secretar citocinas y factores de crecimiento que junto a los fenómenos anteriores provoca una inflamación en el intersticio de la pared arterial.
La médula ósea ictus e infartos
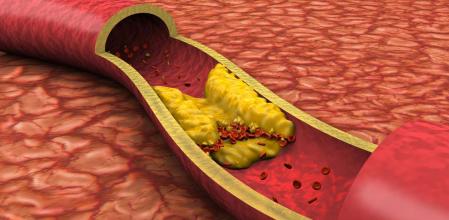
Ilustración de una acumulación de colesterol en una arteria Getty
Las personas con factores de riesgo cardiovascular pero sin síntomas de enfermedad tienen una actividad anómala de la médula ósea que favorece el desarrollo de la aterosclerosis, según un estudio del Centro Nacional de Investigaciones Cardiovasculares (CNIC). El descubrimiento cambia la visión de cómo se originan las enfermedades cardiovasculares y confirma la estrategia de prevenirlas antes de que aparezcan los síntomas.
El estudio se ha basado en datos de 745 personas aparentemente sanas con una media de edad de 50 años. Según los resultados presentados en la revista European Heart Journal, las personas con obesidad, hipertensión, nivel alto de triglicéridos o de azúcar en sangre o con nivel bajo de colesterol HDL producen un exceso de células inflamatorias en la médula ósea.
Estas células inflamatorias dañan los vasos sanguíneos, lo que alimenta un círculo vicioso en el que el daño en las arterias causa inflamación y la inflamación causa daño en las arterias. A falta de medidas de prevención o tratamiento para interrumpir el círculo vicioso, la enfermedad cardiovascular progresa y puede acabar causando ictus o infartos de miocardio.
Esta es la primera investigación que demuestra una relación tan directa entre los factores de riesgo y la activación de la médula ósea.
Los investigadores han evaluado la actividad de la médula ósea de los 745 participantes con la tecnología de imagen PET. Asimismo, se les ha examinado el estado de las arterias con PET y con resonancia magnética. El estudio forma parte del proyecto PESA-CNIC-Santander, iniciado en 2010 y programado hasta el 2029 para comprender cómo se inicia la enfermedad cardiovascular y mejorar su prevención.
Según los investigadores del CNIC, la médula ósea se activa como mecanismo de defensa cuando se acumula colesterol en la pared de las arterias. En una investigación anterior demostraron que las células inmunitarias procedentes de la médula retiran el colesterol de las arterias. Sin embargo, si la acumulación de colesterol es excesiva, el sistema inmunitario se ve desbordado y desencadena una reacción inflamatoria perjudicial.
Aterosclerosis e infección.
Varios gérmenes se han señalado como culpables del desarrollo de las placas de ateroma
Las infecciones actuarían como un factor de riesgo más en el desarrollo de la aterosclerosis. Son muchas las hipótesis que intentan explicar esta asociación 

En la génesis de la aterosclerosis se han involucrado a diferentes microorganismos de entre ellos, el CMV, C. pneumoniae . H. pylori yl Virus del Herpes
Los fenómenos de respuesta inflamatoria frente a cualquier agente infeccioso pueden provocar daño endotelial que precipite la aterogénesis. La formación de inmunocomplejos y la síntesis de citocinas pueden dañar el endotelio directamente.
Modelos animales.
La evidencia del posible papel patogénico del CMV en la aterosclerosis proviene de los experimentos en animales realizados por Fabricant y Hajjar12,28. En los años cuarenta, Paterson y Cottral5 descubrieron una asociación entre la aterosclerosis y el virus de la enfermedad de Mareck. Posteriormente, Fabricant inyectó virus de la enfermedad de Mareck a pollos sanos y les sometió a una dieta rica en colesterol, escogiendo como controles a pollos no infectados pero alimentados con la dieta rica en lípidos. En las autopsias había lesiones aterosclerosas en toda la pared arterial de los pollos infectados, mientras que estas lesiones eran mínimas o inexistentes en los sanos. También observó que estas lesiones eran más extensas en aquellos pollos infectados y alimentados con la dieta rica en colesterol. La vacunación previa de los pollos contra el virus de la enfermedad de Mareck les protegía frente al acúmulo de lípidos en la pared arterial34.
Los experimentos de Hajjar en la década de los ochenta demostraron que la infección de pollos con el virus de la enfermedad de Mareck provocaba un mayor acúmulo de colesterol, ésteres de colesterol, triglicéridos y fosfolípidos en las paredes aórticas12 .
EL H. PYLORI es una bacteria microaerofílica, gramnegativa, de forma espiral, que reside en las células de la mucosa gástrica y que se transmite por un mecanismo fecal-oral. Está presente en prácticamente el 100% de las úlceras duodenales y en el 60% de las úlceras gástricas. La infección por H. pylori se adquiere habitualmente en la infancia y los anticuerpos frente a ella persistirán hasta edades avanzadas de la vida, por lo que la prevalencia de anticuerpos frente a Helicobacter en la población general es muy alta, alrededor del 40% a los 50 años de edad17. 
CITOMEGALOVIRUS (CMV) Cuando infecta una célula se producen cambios en la misma que conducen a su muerte. Cuando estos cambios no se producen se habla de infección abortiva. Experimentalmente esta forma de infección puede conseguirse infectando una célula con una cepa de CMV que tiene tropismo para infectar células de otras especies, por ejemplo, infectar células humanas con CMV obtenido de células de pollo infectadas. La infección abortiva de CMV induce la expresión de los productos de genes inmediatos, como IE2-84, que se unen al p53 inhibiendo su acción y provoca que la célula no finalice el ciclo celular11. La infección abortiva por CMV inhibe el fenómeno de apoptosis necesario para el recambio celular en los tejidos. El CMV parece más relacionado con la aterosclerosis desarrollada tras el trasplante cardíaco, así como con el fenómeno de reestenosis tras una angioplastia coronaria 
HERPES VIRUS .
Se ha demostrado que las infecciones por este virus disminuyen la actividad lisosómica y citoplásmica de la hidrólisis de colesterol, provocando su acumulación en la placa de ateroma12. Parece que la infección por virus de la familia herpes facilita el acúmulo de colesterol en el interior de las células musculares lisas mediante un aumento de la captación de las LDL-ox. Asimismo, las infecciones víricas condicionan un estado procoagulante en el endotelio y el mecanismo más importante sería la exposición al torrente circulatorio del factor tisular, que es un poderoso coagulante33. 
.
CHLAMYDIA PNEUMONIAE
Chlamydia pneumoniae es una bacteria gramnegativa, de crecimiento intracelular. Se transmite a través de las secreciones respiratorias y se cree que persiste en el interior de los macrófagos alveolares. Es el segundo patógeno causante de neumonías atípicas, responsable del 10% de los casos. La primoinfección por C. pneumoniae suele ocurrir en edades tempranas de la vida y las reinfecciones son extraordinariamente frecuentes. Aproximadamente el 50% de los mayores de 50 años tienen serología positiva frente a C. pneumoniae y su prevalencia se correlaciona con la edad, el nivel socioeconómico, el hábito tabáquico y las epidemias periódicas. Es mucho más frecuente en los varones37.
Los resultados de los estudios seroepidemiológicos muestran una asociación fuerte entre serología positiva frente a C. pneumoniae y aterosclerosis, con una OR > 217.La mayoría de ellos están realizados en cardiopatía isquémica clásica (angina de pecho, infarto de miocardio) y algunos en patología cerebrovascular. El trabajo de Saikku, publicado en 19882, marcó la pauta para el desarrollo de trabajos ulteriores. En este estudio, realizado en una población de 40 enfermos con infarto agudo de miocardio, en 30 con cardiopatía isquémica crónica y en 41 controles, cerca del 70% de los pacientes con infarto de miocardio tenía serología positiva frente a C. pneumoniae a títulos >1:128. El 50% de los que padecían cardiopatía isquémica crónica tenían niveles de IgG e IgA significativamente mayores que los controles. No se encontraron diferencias entre los pacientes con infarto y aquellos con angina de pecho. Sin embargo, no todos los trabajos muestran una asociación positiva a pesar de los títulos altos de anticuerpos frente a Chlamydia39. 
Estudios patológicos
Existen numerosas evidencias patológicas que relacionan la infección por C. pneumoniae con la arterioesclerosis de cualquier localización.Thom et al40 encontraron correlación entre los niveles de anticuerpos y la severidad de las lesiones angiográficas. En diferentes estudios de muestras obtenidas por necropsia se ha encontrado la bacteria en las placas de ateroma, pero también en las células musculares lisas y en las células espumosas, mientras que no se evidenciaba en las paredes arteriales sanas17. La mayoría de estos estudios identifican a la Chlamydia por técnicas de inmunohistoquímica o PCR. También ha sido posible detectar la bacteria en muestras in vivo en carótidas, aorta abdominal, arterias periféricas y, recientemente, en válvulas aórticas no reumáticas41,42. En algunos estudios se ha llegado a aislar la bacteria mediante cultivo en líneas celulares.
En trece estudios publicados que demuestran la presencia de C. pneumoniae en muestras patológicas en función de la presencia de ADN, antígenos o cuerpos elementales, la infección local se confirmó en un 52% de las lesiones ateromatosas por sólo un 5% de los controles con arterias sanas17. Aunque ello no significa que en las lesiones haya bacterias viables, en ocasiones se ha conseguido su cultivo en muestras patológicas.
Los mecanismos propuestos para explicar esta asociación han sido diversos. Algunos trabajos encuentran que aquellos con títulos elevados de anticuerpos frente a Chlamydia presentan concentraciones plasmáticas elevadas de proteína C reactiva y de fibrinógeno43,44. Asimismo, la infección podría desencadenar la liberación de mediadores químicos por parte de los macrófagos (interleucina 1, interferón *, FNT-*) que estimulan la respuesta inflamatoria.
Al igual que con H. pylori, el daño endotelial en la infección por Chlamydia puede desencadenarse por una reacción cruzada frente a determinantes antigénicos similares, en este caso las HSP6045.
A pesar de estas evidencias, no es posible establecer si la infección por C. pneumoniae desencadena el proceso aterosclerótico o, por el contrario, acelera este proceso una vez iniciado por los factores de riesgo ateroscleróticos clásicos.
Ensayos de intervención terapéutica Ensayos de intervención terapéutica
Un estudio experimental en ratas en las que se realizaba un trasplante cardíaco sugiere que la administración de ganciclovir a ratas portadoras de CMV disminuye los efectos de la vasculopatía del trasplante35.
Algunos estudios realizados en receptores humanos de trasplante cardíaco infectado por CMV sugieren que la administración profiláctica de ganciclovir reduce la incidencia de enfermedad vascular relacionada con el virus36, pero hasta la fecha no hay trabajos que permitan establecer la eficacia de ganciclovir en la reducción de la incidencia de aterosclerosis.
Es extraordinariamente difícil distinguir entre infecciones agudas o crónicas y, dentro de estas últimas, entre reagudizaciones y cronicidad. Esto es debido a que los métodos de detección no son fiables y a que las formas clínicas son superponibles. Existen diversos métodos de identificación en el laboratorio. El método de fijación de complemento no es útil, puesto que no diferencia entre las distintas especies de Chlamydia debido a que el lipopolisacárido de la cubierta es muy similar en todas ellas. Las técnicas de inmunofluorescencia son las más utilizadas en los estudios, pero son muy complejas y requieren experiencia a la hora de interpretarlas. Las técnicas mediante PCR no están muy extendidas y su sensibilidad no es muy buena. El cultivo es extraordinariamente difícil, requiere medios celulares y su sensibilidad es menor que las técnicas serológicas.
Otro problema añadido es la falta de unanimidad a la hora de definir los títulos de anticuerpos. Si bien existe consenso en que la infección aguda se caracteriza por un aumento en los títulos de IgM seguido más tarde de una pequeña elevación en los títulos de IgG o bien una seroconversión de al menos un aumento de cuatro veces los títulos de IgG, no está clara la distinción entre infección crónica persistente y reinfección. Se ha dado valor a la elevación persistente de IgA específica junto con aumento de IgG como marcador de infección crónica debido a que la vida media de la IgA es más corta que la de IgG38
Hasta la fecha sólo existen dos grandes estudios de intervención terapéutica en humanos, aunque actualmente hay varios trabajos en desarrollo que intentan demostrar un efecto beneficioso del tratamiento con antibióticos. El primer estudio de Gurfinkel et al50 (estudio ROXIS) evaluó el posible papel protector de la Roxitromicina en la prevención de nuevos episodios isquémicos. Seleccionaron 202 pacientes (la mayoría varones) que habían sufrido un infarto agudo de miocardio o un episodio de angina inestable. Se distribuyeron aleatoriamente en dos grupos, para recibir roxitromicina, 150 mg dos veces al día durante treinta días, o placebo. El período de seguimiento del estudio fue de seis meses y como eventos finales se estimaron la muerte por cardiopatía isquémica, nuevo episodio de infarto o isquemia recurrente. El análisis de los eventos por separado demostró una cierta tendencia beneficiosa, pero no estadísticamente significativa. Cuando se analizaron los tres eventos finales juntos se obtuvo una tasa significativamente menor de sucesos en el grupo activo que en el placebo (1% frente a 10%, p = 0,032).
El otro gran estudio realizado por Gupta et al51 incluyó a 213 varones que habían sufrido un infarto de miocardio. Los enfermos fueron divididos en tres grupos según los títulos de anticuerpos frente a C. pneumoniae. Un grupo con serología negativa, otro con títulos entre 1/8 y 1/32 y el último con títulos superiores o igual a 1/64. A este último grupo se les subdividió aleatoriamente en tres subgrupos: unos recibieron azitromicina 500 mg/día durante tres días, otros recibieron dos tandas de azitromicina de tres días, separados entre sí tres meses, y el último grupo recibió placebo. El período de seguimiento fue de 18 meses. El primer objetivo del trabajo era comparar la reducción de los títulos de anticuerpos. Los objetivos secundarios eran analizar la disminución de episodios coronarios: muerte de causa cardíaca, infarto de miocardio no fatal, angina inestable e infarto no-Q o angina inestable susceptible de coronariografía. Después de seis meses un 43% de los que recibieron antibiótico redujeron sus títulos de anticuerpos hasta 1/16 frente a un 10% de los que recibieron placebo (p < 0,02). Analizando los objetivos secundarios encontraron que en el grupo tratado con azitromicina el riesgo de acontecimientos clínicos era cuatro veces inferior a los no tratados (p = 0,03).
Conclusiones
Es posible que los procesos infecciosos puedan intervenir en la patogenia del fenómeno aterosclerótico. La hipótesis de respuesta a la lesión desarrollada por Ross permite apreciar la aterosclerosis como un fenómeno inflamatorio. Esta hipótesis, hoy globalmente aceptada, nos ofrece posibles vías de actuación de agentes infecciosos perpetuando o provocando el fenómeno inflamatorio que va a desembocar en la placa de ateroma. Hasta la fecha las mayores evidencias favorecen al CMV y Chlamydia pneumoniae. Por el contrario, hay escasas evidencias que apoyen el papel de Helicobacter pylori en el desarrollo de aterosclerosis.
BIBLIOGRAFÍA
[1] Shattuck lecture. Cardiovascular medicine at the turn of the millennium: triumphs, concerns, and opportunities. N Engl J Med 1997; 337:1.360-1.369.
[2] Serological evidence of an association of a novel Chlamydia, TWAR, with chronic coronary heart disease and acute myocardial infarction. Lancet 1988; 2(8618) 983-986.
[3]Frothingham C.. The relationship between acute infectious diseases and arterial lesions.. Arch Intern Med, 8 (1911), pp. 153-162
[4] Woodhause PR, Khaw KT, Plummer M, Foley A, Meade TW.. Seasonal variations of plasma fibrinogen and factor VII activity in the elderly: winter infections and death from cardiovascular disease.. Lancet, 343 (1994), pp. 435-439
[5] Paterson JC, Cottral GE.. Experimental coronary sclerosis: lymphomatosis as a cause of coronary sclerosis in chickens.. Arch Pathol, 49 (1950), pp. 699-709
[6] Phlogose and thrombose in gefassystem, gesammelte abhandlungen zur wissenscharftlichen medicin. Frankfurt-am-Main: Meidinger Sohn; 1856; 458.
[7] Ross R, Glomset JA.. The pathogenesis of atherosclerosis (first of two parts).. N Engl J Med, 295 (1976), pp. 369-377
http://dx.doi.org/10.1056/NEJM197608122950707 | Medline
[8] Ross R.. Mechanisms of disease: atherosclerosis. An inflamatory disease.. N Engl J Med, 340 (1999), pp. 115-126
[9] Aterosclerosis e infecci??n. ??Estamos olvidando algo? Infecci??n 1999; 98.
[10] The pathogenesis of atherosclerosis. En: Braunwald E, ed. The heart. A textbook of cardiovascular medicine (5.a ed). Philadelphia: WB Saunders Co, 1997; 1.105-1.121.
[11]Speir E, Modali R, Huang ES, Leon MB, Shawl F, Finhel T, Epstein SE.. Potencial role of human cytomegalovirus and p53 interaction in coronary restenosis.. Science, 256 (1994), pp. 391-394
[12] Herpes simplex virus infection in human arterial cells: implications in atherosclerosis. J Clin Invest 1987; 80:1.317-1.321.
[13] Human cytomegalovirus increases modified low density lipoprotein uptake and scavenger receptor mRNA expression in vascular smooth muscle cells. J Clin Invest 1996; 98:2.129-2.138.
[14] Infection and inflammation as risk factors for myocardial infarction. Eur Heart J 1993; 14 (suppl K):12-16.
[15]Correlation between antibodies to heat shock protein 65 and coronary atherosclerosis: possible role of Helicobacter pylori infection. Eur Heart J 1996; 18 (suppl):231.
[16]Epstein SE, Zhou YF, Zhu J.. Infection and atherosclerosis. Emerging mechanistic paradigms.. Circulation, 100 (1999), pp. e20-e28
[17] Chronic infections and coronary heart disease: is there a link? Lancet 1997; 350:430-436.
[18] High seroprevalence of Helicobacter pylori infection in coronary heart disease (carta). Lancet 1995; 346:310.
[19] Helicobacter pylori seropositivity in myocardial infarction (carta). Lancet 1995; 345:1.380.
[20] Association of Helicobacter pylori infection with coronary heart disease (carta). BMJ 1996; 312:251.
[21] Helicobacter pylori and the heart. Metanalysis of the literature. Gastrointest Intern 1997; 10:8.
[22] De Luis DA, Aller R, Boixeda de Miquel D.. Helicobacter pylori y patología cardiovascular. De nuevo la hipótesis inflamatoria de la arteriosclerosis.. An Med Intern, 17 (2000), pp. 115-117
[23] Ossei-Gerning N, Moayyedi P, Smith S, Braunholtz D, Wilson J, Axon A, Grant P.. Helicobacter pylori infection is related to atheroma in patients undergoing coronary angiography.. Cardiovasc Res, 35 (1997), pp. 120-124
[24] Correlation between gastric infection with Helicobacter pylori and plasma levels of fibrinogen, plasminogen activator (PAI) and von Willebrand factor (vWF) antigen. Gastroenterology 1996; A 90.
[25] Changes in plasma fibrinogen levels after eradication of H. pylori infection in patients affected by coronary heart disease. Gastroenterology 1997; A 313.
[26] Fibrinogen: a link between chronic and coronary heart disease. Lancet 1994: 343:1.634-1.636.
[27] Sung JJ, Sanderson JE.. Hyperhomocysteinaemia, Helicobacter pylori and coronary heart disease.. Heart, 76 (1996), pp. 305-307
[28] Fabricant CG, Fabricant J, Litrenta M, Minick C.. Virus-induced atherosclerosis..
J Exp Med, 148 (1978), pp. 335-340
[29] Association between prior cytomegalovirus infection and the risk of restenosis after coronary atherectomy. N Engl J Med. 1996; 335:624-630.
[30] Olivari M, Kubo S, Braunlin E, Bolman R, Ring W.. Five year experience with triple drug immunosuppressive therapy in cardiac transplantation.. Circulation, 82 (1990), pp. 276-280
[31] Cytomegalovirus infection is associated with cardiac allograft rejection and atherosclerosis. JAMA 1989; 261:3.561-3.566.
[32] The presence of cytomegalovirus nucleic acids in arterial walls of atherosclerotic and non atherosclerotic patients. Am J Pathol 1989; 134:1.151-1.157.
[33] Infection of vascular endothelial cells with herpes simplex virus enhances tissue factor activity and reduces thrombomodulin expression Proc Nat Acad Sci USA 1990; 87: 7.0
[34] Minick CR, Fabricant CG, Fabricant J, Litrenta MM.. Atherosclerosis induced by infection with a herpes virus.. Am J Pathol, 96 (1979), pp. 673-706
[35] Cytomegalovirus infection enhanced cardiac allograft vasculopathy is abolished by DHPG prophylaxis in the rat. Circulation 1997; 95:2.614-2.616.
[36] A controlled trial of ganciclovir to prevent cytomegalovirus disease after heart transplantation. N Engl J Med 1992; 326: 1.182-1.186.
[37] Pathogenetic mechanisms and epidemiology of Chlamydia pneumoniae. Eur Heart J 1993; 14 (suppl K):57-61.
[38] Wong YK, Gallagher PJ, Ward ME.. Chlamydia pneumoniae and atherosclerosis.. Heart, 81 (1999), pp. 232-238
[39] Kark JD, Leinonen M, Paltied O, Sailku P.. Chlamydia pneumoniae and acute myocardial infarction in Jerusalem. Int J Epidem, 26 (1997), pp. 730-738
[40] Thom DH, Wang SP, Grayston JT, Siscovick DS, Stewart DK, Kronmal RA, et al.. Chlamydia pneumoniae strain TWAR antibody and angiographically demonstrated coronary artery disease. Arterioscler Thromb, 11 (1991), pp. 547-551
[41] Ramírez JA.. Isolation of Chlamydia pneumoniae from the coronary artery of a patient with coronary atherosclerosis.. Ann Int Med, 125 (1996), pp. 979-982
[42] Juvonen J, Juvonen T, Laurila A, Ala Karpa H, Lounatmae.k, Surcel HM, et al.. Demonstration of Chlamydia pneumoniae in the walls of abdominal aortic aneurysms.. J Vasc Surg, 25 (1997), pp. 499-505
[43] Patel P, Mendall MA, Carrington D, Strachan DP, Leatham E, Molineaux N, et al.. Association of Helicobacter pylori and Chlamydia pneumoniae infections with coronary heart disease and cardiovascular risk factors.. BMJ, 311 (1995), pp. 711-714
[44] C reactive protein and its relation to cardiovascular risk factors: a population based cross sectional study BMJ 1996; 312:1.0
[45] Autoantibodies against Heat Shock Protein 60 mediate endothelial cytotoxicity. J Clin Invest 1995; 96:2.569-2.577.
[46] Moazed TC, Kuo C, Grayston JT, Campbell LA.. Murine models of Chlamydia pneumoniae infection and atherosclerosis..
[47] Fong IW, Chiu B, Viira E, Fong MW, Jang D, Mahony J.. Rabbit model of Chlamydia pneumoniae infection.. J Clin Microb, 35 (1997), pp. 48-52
[48] Muhlestein JB, Anderson JL, Hammond EH, Zhao L, Trehan S, Schwobi EP, et al.. Infection with Chlamydia pneumoniae accelerates the development of atherosclerosis and treatment with azithromycin prevents it in a rabbit model..Circulation, 97 (1998), pp. 633-636
[49] Moazed TC, Kuo C, Patton DL, Grayston JT, Campbell LA.. Experimental rabbit models of Chlamydia pneumoniae infection.. Am J Pathol, 148 (1996), pp. 667-676
[50] Gurfinkel E, Bozovich G, Daroca A, Beck E, Mautner B.. Randomized trial of roxitromycin in non-Q wave coronary syndromes: ROXIS pilot study.. Lancet, 350 (1997), pp. 404-407
[51] Gupta S, Leatham EW, Carrington D, Mendall MA, Kaski JC, Camm AJ.. Elevated Chlamydia pneumoniae antibodies, cardiovascular events and azithromycin in male survivors of myocardial infarction.. Circulation, 96 (1997), pp. 404-407
JOSEP CORBELLA BARCELONA 22/03/2022 11:48
Final del formulario

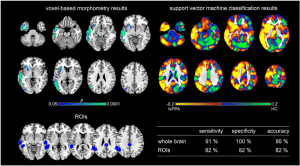
 Víctor Montal, investigador del IIB Sant Pau y primer firmante Foto: IIB SANT PAU.
Víctor Montal, investigador del IIB Sant Pau y primer firmante Foto: IIB SANT PAU.
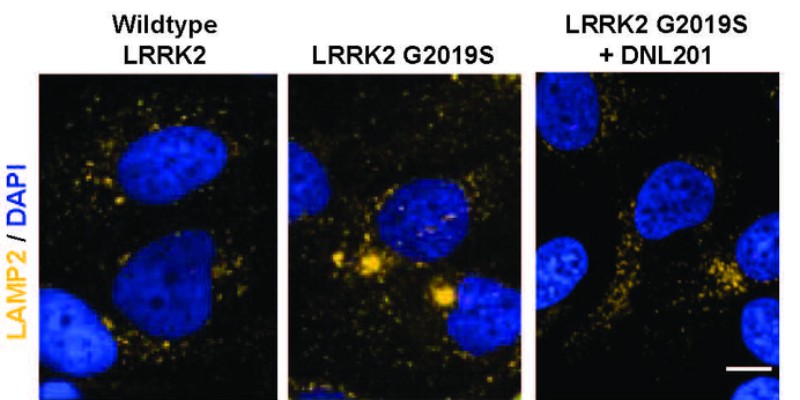 Células humanas que expresan LRRK2, a las 72 horas del tratamiento; la estructura de los lisosomas se marca en amarillo. Foto: STM
Células humanas que expresan LRRK2, a las 72 horas del tratamiento; la estructura de los lisosomas se marca en amarillo. Foto: STM
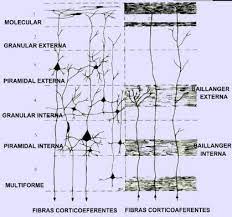 neuronales que están implicados en funciones corporales vitales como la r
neuronales que están implicados en funciones corporales vitales como la r  espiración están bien desarrollados.
espiración están bien desarrollados.