HISTORIA DE LA ENFERMEDAD DE PARKINSON DURANTE LOS DOS ÚLTIMOS SIGLOS

“Essay on the shaking palsy “,es el nombre que le dio James Parkinson a esta enfermedad, Y pese a que es muy antigua, este nombre persiste . Fue publicado en 1817, y su descripcion fue y es magistral1

Esta enfermedad es más antigua de lo que inicialmente se creía; al parecer, culturas ancestrales tenían conocimiento de ella,2 incluso, el mismo Parkinson reconoció que otros autores que le precedieron ya habían descrito signos de la enfermedad.3 Gaceta Médica de México. 2018;154 720 Uno de los datos más antiguos se encuentra en papiros egipcios correspondientes a la Dinastía XIX (1500-1200 a. C.), donde se evoca la sialorrea de los pacientes con esta enfermedad al detallar a un rey al cual “la edad había aflojado su boca” y “escupía continuamente”.4 La Ayurveda (āyuh: ‘duración de la vida’, veda ‘conocimiento’), o antiguo sistema de medicina tradicional de la India, fue publicada alrededor del 1000 a. C. y en ella se menciona a la kampavata (kampa: temblor), enfermedad que se caracteriza por escaso movimiento, exceso de saliva, somnolencia y “mirada reptiliana”.5 Se conoce también que en el Tratado de medicina tradicional china (Nei- Jing), que data de alrededor de 800 a. C., se describen personas con temblor cefálico y de manos, cuya explicación era atribuida de igual forma a la edad.4 En el Antiguo Testamento de la Biblia existen algunas citas interesantes, ejemplo de ello es Eclesiastés 12:3 que dice: “Un día temblarán los guardianes de la casa, y se encorvarán los hombres de batalla; se detendrán las molenderas por ser tan pocas, y se apagarán los que miran a través de las ventanas”, describiéndose la postura y el temblor que podría hacer referencia a EP. Mucho tiempo después, Galeno de Pérgamo, considerado uno de los médicos más influyente de Europa, definió el temblor y pudo distinguirlo entre el de acción y reposo en sus observaciones.5 Lamentablemente, durante siglos desapareció la información relativa a la EP, quizá debido a las numerosas batallas territoriales en las cuales se vio sumergida Europa, que disminuyó la esperanza de vida de los pobladores de esos tiempos, por lo que se podría sospechar que la edad de muerte impedía que la senectud fuera un aspecto común dentro de la sociedad y por ello, probablemente, algunas enfermedades neurodegenerativas pudieron no ser tan frecuentes.6 Las posteriores descripciones que parecen relacionarse con la EP vuelven a emerger a partir del siglo XVII: en 1641, el famoso doctor Nicolaes Tulp retomó el término temblor. Este personaje fue tan importante en su época que le mereció uno de los retratos más conocidos de la historia realizado por el pintor holandés Rembrandt. De igual forma, Silvio de La Boe habló de las características del temblor de acción y en reposo, descripción por la que fue señalado por Parkinson en su ensayo.7 Otras figuras, como Leonardo Da Vinci, Shakespeare o Rembrandt y médicos de la talla de Johanness Baptiste Sagar, Boissier de Sauvages, John Hunter y Chomel hicieron también referencia al temblor y otros trastornos motores en sus obras. En 1758, Gaubius refirió haber observado a “un hombre capaz de correr, pero no de andar”, con lo que aludió a las alteraciones de la marcha que se observan en esta patología.5 Finalmente, en 1817 se publicó el ensayo de James Parkinson, donde por primera vez se recogieron específicamente detalles de la enfermedad, extraídos de la observación de seis pacientes. En ese momento se dio a conocer que el temblor, la bradicinesia y la inestabilidad postural son los signos más importantes de esta entidad. Parkinson indicó que se trataba de un “movimiento tembloroso involuntario… en partes que no están en actividad”, además destacó la “propensión a flexionar el tronco hacia adelante”.3 Al describir la historia natural de la enfermedad, con una redacción claramente coherente y sencilla, reveló que el inicio de la enfermedad era lentamente progresivo, que comenzaba de forma unilateral y que en poco tiempo comprometía el lado contralateral del cuerpo, con posterior compromiso de la posición erecta al caminar y desarrollo de anteropulsión de la marcha. Mencionó que a medida que el tiempo avanzaba, el paciente sentía que los movimientos perdían precisión y los realizaba con gran dificultad. Las caídas se hacían más notorias por la incapacidad de elevar fácilmente las piernas y el lenguaje se volvía inteligible, por lo que el paciente requería la presencia permanente de un cuidador. Quizás una de sus consideraciones con más significación en la actualidad fue la descripción de algunos síntomas no motores de la enfermedad, por ejemplo, las alteraciones del sueño, el estreñimiento, la sialorrea y las alteraciones de los esfínteres. James Parkinson encontró numerosos detractores, aunque finalmente las observaciones directas de afamados médicos de la época resultaron ser útiles para refinar los datos diagnósticos, como sucedió con Jean-Martin Charcot, quien no pudo obtener una versión del referido “Essay on the shaking palsy” traducido al francés sino hasta 1881, gracias a la tarea de traducción que él mismo encomendó a sus estudiantes, luego de lo cual emitió el siguiente comentario: Esta es una definición descriptiva y vívida que es correcta en muchos casos, la mayoría de hecho, y siempre tendrá la ventaja sobre otros de haber sido el primero, pero se equivoca por ser demasiado general. Charcot es quien relacionó por primera vez la rigidez con la enfermedad y señaló que es el signo cardinal de la misma, logrando diferenciarla de la espasticidad.8 De esta manera, parecía que se había Arredondo-Blanco K, et al.: 200 años de Parkinson 721 alcanzado el conocimiento para describir la enfermedad, pero bastaría que los siglos transcurrieran para evidenciar que otras manifestaciones, actualmente denominadas como síntomas no motores, pertenecen a la misma entidad.
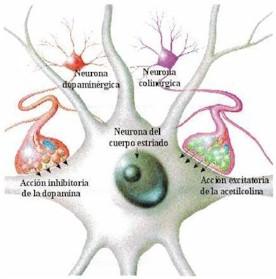
Historia de la anatomía e histopatología Después de la descripción clínica de James Parkinson en 1817, así como de los escritos de Jean Martin Charcot titulados “Leçon sur le maladies du systeme nerveux”, uno de los acontecimientos más importantes para la compresión de la EP fue el descubrimiento de la pérdida neuronal en la pars compacta de la sustancia nigra. Esta pérdida fue identificada como la lesión patológica que caracteriza a la EP, lo cual se mantiene hasta la fecha.9 A principios del siglo XX se comenzó a investigar sistemáticamente los mecanismos patogénicos de la enfermedad. En 1913, el patólogo Friederich Lewy describió las inclusiones citoplásmicas de alfa-sinucleína localizadas en neuronas del núcleo motor dorsal del nervio vago y del núcleo basal de Meynert. Estas inclusiones recibieron posteriormente la denominación de cuerpos de Lewy.10 Seis años más tarde, en 1919, Konstantine Trétiakoff dio nombre a la sustancia nigra, detalló su localización y asoció la pérdida de neuronas en esta zona como parte de los hallazgos de la EP.11 En 1960, con el uso de microscopia electrónica, se demostró la estructura interna filamentosa de los cuerpos de Lewy12 y es hasta 2003 cuando Braak et al. publicaron los resultados de los estudios seriados que realizaron en cerebros post mortem, donde evidenciaron la presencia de cuerpos de Lewy en diversas zonas del sistema nervioso, diseñando además un modelo para estadificar la progresión de la enfermedad y su relación con los síntomas motores y no motores.13 En la década de 1980 se reconocieron los efectos neurotóxicos en un grupo de adictos a una “nueva heroína sintética”. Estos sujetos desarrollaron secundariamente un parkinsonismo relacionado con lesión en la sustancia nigra. Fue así como se identificó al MPTP (1-metil-4-fenil-1,2,3,6 tetrahidropiridina) como el agente causal; posteriormente este hallazgo dio origen a los modelos experimentales de EP en animales, que hasta la fecha continúan siendo de gran utilidad.14 En la actualidad se ha descrito no solo la presencia de depósitos de alfa-sinucleína, sino también de proteína tau y de beta amiloide, lo que ha llevado al uso del término de proteinopatías, donde probablemente la patología predominante es la que distinga fenotípicamente a la EP de otras enfermedades como taupatías, Alzheimer o demencia frontotemporal.13 En la última década se ha documentado una transmisión célula a célula de la alfa-sinucleína, al menos en modelos animales y celulares, lo que ha llevado a considerar la posibilidad de que la EP sea un trastorno similar a las enfermedades priónicas.15 Este concepto, de comprobarse, no solo será de gran relevancia para la comprensión de la fisiopatología de la enfermedad, sino también ofrecerá la oportunidad de desarrollar terapia molecular para modificar la progresión de la enfermedad.16 Historia de la genética de la enfermedad de Parkinson Durante décadas se consideró que la EP era una patología de presentación exclusivamente esporádica, ya que aun cuando existían evidencias de un componente familiar, no se disponían de técnicas científicas que permitieran comprobar tal fenómeno. Sin embargo, la evolución tecnológica permitió que este paradigma cambiara. Las primeras investigaciones genéticas sobre esta patología se iniciaron a finales de la década de 1990, en las que se asumió que al menos un porcentaje de los pacientes con EP tiene una causa genética a pesar de la discordancia monocigótica en gemelos que demostraban los estudios de esa época. En 1997, al estudiar cuatro grandes familias con EP se descubrió que una mutación en el gen que codifica la alfa sinucleína ocasiona una forma de herencia dominante de la EP17 y se logró establecer una relación entre los acúmulos de alfa sinucleína y los cuerpos de Lewy.18 Como consecuencia, en los siguientes años esta proteína se convirtió en el blanco de numerosos estudios de investigación. Los parkinsonismos juveniles también se beneficiaron de las técnicas de estudio de material genético. En 1998 se descubrió que mutaciones en la proteína ahora denominada parkina eran la causa de una forma de presentación temprana de la enfermedad.19 En 2003 se logró identificar mutaciones en el gen DJ1 y se asoció como causa de parkinsonismo de inicio temprano.20 PINK 1 fue descubierto en 2004, sugiriéndose como la segunda causa más común de parkinsonismo autosómico recesivo.21 En este mismo año, se reconoció el LRRK2 mediante el mapeo de recombinación de alta resolución y la secuenciación de Gaceta Médica de México. 2018;154 722 genes candidatos en 46 familias, con lo que se iniciaron largos estudios de investigación.22 El crecimiento en el conocimiento genético ha permitido entender la fisiopatología y fenomenología de la EP, además de generar interés en estrategias como terapia génica y reprogramación celular para el manejo de la enfermedad a futuro. A la fecha se conocen 17 genes diferentes considerados como causales, ya sea de forma autosómica dominante o recesiva; adicionalmente se ha identificado un gran número de genes, que, aunque no causales, son considerados de riesgo. Historia del tratamiento de la EP Existen descripciones en la literatura que datan de casi 3000 años atrás, desde la medicina ayurvédica. El tratado clásico médico hindú hace referencia sobre la enfermedad de kampavata, la cual se trataba con un conjunto de remedios vegetales derivados de plantas del género mucuna (Mucuna pruriens), con alto contenido de levodopa obtenida al triturar las semillas. La mucuna es una liana de las selvas tropicales de Asia y América, por lo cual se utilizó también en la región amazónica. En la medicina ayurvédica se daba a los enfermos un brebaje de polvo de semillas de mucuna cocido en leche de vaca, que mejoraba el temblor, la bradicinesia, la rigidez y los calambres, si bien se observaba la presencia de sialorrea. Recientemente se ha demostrado que cada dosis de polvo de semillas contiene alrededor de 200 mg de levodopa.23 En escritos de medicina herbolaria antigua se encuentra información sobre tratamientos contra el temblor con semillas de estramonio, por su efecto anticolinérgico. Las habas y otras legumbres exóticas también contienen levodopa y se utilizan desde hace varios siglos en el tratamiento de la EP.24,25 Por otra parte, desde 1887 Wilhelm Erb introdujo el uso terapéutico de la escopolamina y preparaciones similares fueron utilizadas por sus efectos farmacológicos anticolinérgicos en la década de 1950. Ordestein, discípulo de Charcot, en su tesis médica mencionó el uso de alcaloides de la belladona para controlar el temblor.26 Los tratamientos con efectos anticolinérgicos de Charcot proporcionaron las bases farmacológicas de los modernos medicamentos antiparkinsonianos. La oportunidad histórica de analizar los tratamientos primarios de la enfermedad se encontró con el hallazgo de cartas en la biblioteca personal de este eminente neurólogo. Estas cartas cubren un periodo de 15 meses, de enero de 1863 a marzo 1864.27 William Gowers, contemporáneo de Charcot, utilizaba estrategias terapéuticas similares. Adicionalmente, Gowers describió el efecto negativo del estrés, el cansancio físico y mental, recomendando una vida tranquila;28 para el temblor indicaba hiosciamina, arsénico y cáñamo, así como la combinación de Cannabis y opio.29 En 1910, en un laboratorio londinense, fue sintetizada por primera vez la dopamina por los investigadores George Barger y James Ewens. En el mismo laboratorio, Henry Dale le otorgó el nombre de dopamina. Cabe mencionar que la levodopa, precursor de la dopamina, fue sintetizada químicamente por Casimir Funk en 1911.30 Entre 1950 y 1960, Arvid Carlsson, ganador del premio Nobel de Fisiología y Medicina en 2000, cambiaría el rumbo del tratamiento de la EP con sus trabajos en relación con la dopamina. Al investigar los cambios vasculares generados por la reserpina en conejos observó que el efecto adverso generado por esta era la acinesia. Después de arduos trabajos de investigación, logró revertir este efecto con uso de la D, L DOPA (L-3,4 dihidroxifenilalanina.31 Posteriormente se describió que en el estriado la depleción de la dopamina es la responsable de la presencia de un efecto acinético mediado por reserpina. El austriaco Oleh Hornykiewicz, ganador del premio Wolf de medicina en 1979, fue el responsable de esta notable descripción.32,33 Hornykiewicz fue el primero en proponer el uso de levodopa para el tratamiento de la enfermedad.34 Uno de los experimentos más significativos realizados en la farmacología de la levodopa estuvo a cargo de Kathleen Montagu, con estudios en animales. En noviembre de 1957 descubrió la dopamina intracelular y su distribución en el encéfalo, además, notó que la levodopa incrementa los niveles de catecolaminas en el cerebro. De igual forma, en 1958, Montagu describió que la reserpina provoca depleción de dopamina y que la levodopa restaura sus niveles.35 En ese mismo periodo, Hermann Blaschko, en el Laboratorio de Fisiología de la Universidad de Cambridge, propuso que la levodopa y la dopamina son metabolitos intermedios en las biosíntesis de catecolaminas.36 En esa misma época, Peter Holtz descubrió la enzima dopamina descarboxilasa y documentó que la levodopa es sintetizada a dopamina a través de esta enzima.37 En 1967, George Cotzias evidenció la eficacia y seguridad del uso de levodopa en personas con EP, en Arredondo-Blanco K, et al.: 200 años de Parkinson 723 dosis de 4 y 8 g al día; como consecuencia de este ensayo se introdujo la levodopa en el tratamiento de la EP en conjunto con la suma de los inhibidores de la dopamina dexcarboxilasa, que actualmente es uno de los fármacos más utilizados y con mayor beneficio en los síntomas motores.38 La inclusión de las dosis adecuadas de levodopa fue un logro innovador en el manejo de los síntomas de la enfermedad.39 Sin embargo, poco tiempo después se evidenció que su uso prolongado ocasiona complicaciones motoras,40 lo que obligó a buscar otras alternativas distintas para el tratamiento farmacológico.41 Por otra parte, Kendal B. Corbin en 1949 demostró que el trihexifenidilo, un anticolinérgico, es útil en el control de los síntomas motores de la EP,42 prolongando la utilización de estos medicamentos. Un progreso importante en materia de tratamiento farmacológico fue el desarrollo de los agonistas de la dopamina, los inhibidores de las enzimas monoaminooxidasa B y los inhibidores de la catecol-o-metiltrasferasa. Estos medicamentos fueron inicialmente aprobados para su uso en humanos en las décadas de 1970 y1980. La apomorfina fue el primer agonista de la dopamina sintetizado en el siglo XIX, sin embargo, data desde la era de los mayas, quienes en rituales religiosos empleaban un extracto de la raíz y el tronco de una planta acuática (Nymphaea ampla y Nymphaea caerulea) con propiedades afrodisiacas y alucinógenas, y que posteriormente se descubriría que contiene apomorfina.43,44 En 1869, Matthienssen y Wrigth observaron que al deshidratar la morfina con ácido clorhídrico se obtenía una sustancia, que llamaron apomorfina; en ese entonces comenzó su uso terapéutico, inicialmente como antiemético.45,46 En 1884 se propuso para el tratamiento de pacientes con parkinsonismo,47 pero no fue hasta 1951 que Schwab et al. reportaron que la apomorfina mejoraba dramáticamente, pero en forma temporal, los síntomas que presentaban los pacientes con EP.48 Posteriormente, en 1965, Ernst puntualizó la similitud estructural entre la apomorfina y la dopamina.49 En 1970, Cotzias et al. reevaluaron la utilización de apomorfina y reportaron efectividad antiparkinsoniana, sin embargo, la toxicidad y la necesidad de la administración por vía parenteral limitó su utilidad.50 Tiempo después se intentó el uso de apomorfina por vía oral,51 pero esta práctica fue abandonada por la presencia de hiperazoemia en terapias a largo plazo. A mitad de la década de 1980, Andrew Lees y Gerald Stern demostraron que el uso por vía subcutánea mediante una bomba de infusión mejoraba en 50 % el periodo off en los pacientes con EP.52 Las dificultades técnicas de la vía de administración y los efectos adversos limitaron el uso de la apomorfina, pero a su vez promovió el uso de otros agonistas dopaminérgicos. Inicialmente se utilizaron agonistas dopaminérgicos derivados de la ergotamina (pergolide, bromocriptina, cabergolida, lisuride), los cuales cayeron en desuso debido a sus efectos adversos (náuseas, somnolencia, alucinaciones); el uso prolongado está relacionado con mayor riesgo de fibrosis pleural, pericárdica, retroperitoneal y fibrosis de la válvula cardiaca.53-55 Fue entonces cuando el tratamiento de la EP se enfocó al uso de agonistas dopaminérgicos no ergotamínicos, como el pramipexol y el ropirinol.56 Aun cuando su efecto sintomático es menor que el de la levodopa, su principal indicación surgió por la necesidad de disminuir el riesgo de complicaciones motoras en etapas tardías de la enfermedad; los problemas derivados han sido motivo suficiente para crear opciones farmacológicas que generen una estimulación dopaminérgica continua, la cual disminuye la pulsatilidad en los niveles de estimulación dopaminaérgica. La rotigotina, agonista dopaminérgico de aplicación transdérmica diaria, se comenzó a utilizar en este siglo XXI.57,58 De igual forma se desarrollaron presentaciones de liberación prolongada tanto de ropinirol como de pramipexol.59 Actualmente la infusión subcutánea de apomorfina se considera un tratamiento de avanzada para pacientes con enfermedad complicada.60 A principios de 2010 se describieron los síntomas asociados con la enfermedad, los síntomas no motores, con múltiples tratamientos para su control y así mejorar la calidad de vida de los pacientes. Aún continúa la búsqueda de nuevas alternativas y el perfeccionamiento de las opciones existentes. Otra estrategia terapéutica involucraba el uso de inhibidores de enzimas específicas implicadas en el metabolismo de la dopamina, como la monoaminooxidasa B (MAO-B). La inhibición de esta enzima incrementa la concentración de dopamina endógena y secundariamente reduce los síntomas de la EP. El primer inhibidor de la MAO-B utilizado en la EP fue la selegilina (inicialmente conocida como deprenil) y más recientemente la rasagilina.61 Otro medicamento relevante es la amantadina, aprobada en 1976 como antiviral, pero su uso para la EP se basó en un descubrimiento fortuito como agente antiparkinsoniano. Schwab notó un inesperado beneficio en la severidad del temblor, marcha y bradicinesia en pacientes con Gaceta Médica de México. 2018;154 724 EP y en pacientes con parkinsonismo posencefalítico.62 En la actualidad, el desarrollo de nuevos terapias con efecto neuroprotector se dirige hacia los anticuerpos monoclonales.

Terapia quirúrgica en la enfermedad de Parkinson En 1908, el cirujano Victor Alexander Haden Horsley y el ingeniero Robert Henry Clarke, en Londres, Inglaterra, fueron los pioneros en crear el equipo de estereotaxia,63 lo que sería un parteaguas en la cirugía de EP. Después de 1930, pero antes de la Segunda Guerra Mundial, los avances en neurocirugía se encontraban enfocados en realizar lesiones en las vías corticoespinales en distintos niveles para tratar de corregir el temblor, sin embargo, las secuelas eran catastróficas. Meyer describió los efectos que se producen con lesiones en el caudado, globo pálido y en el núcleo lenticular, demostrando que el temblor y la rigidez secundarias a la EP mejoran con estos procedimientos quirúrgicos, sin embargo, la mortalidad y morbilidad eran altas.64 A la par, Bucy, Case, y Klemme se enfocaron en la cirugía ablativa, con la que se lesionaba la corteza cerebral para el tratamiento del temblor parkinsoniano; la secuela de este tipo de cirugía fue la hemiparesia, por lo cual fue abandonada.65,66 Alrededor de 1939 a 1940, con la presencia de los conflictos bélicos a nivel internacional, la ciencia tuvo un camino intrincado. Las hostilidades entre países terminaron en 1945, pero fue hasta 1947 que se aprobaron las técnicas neuroquirúrgicas y la estereotaxia se comenzó a utilizar en humanos para efectuar procedimientos lesionales principalmente en el tálamo y el globo pálido.67 A comienzo de la década de 1950, Cooper, durante un procedimiento quirúrgico en un paciente con parkinsonismo, lesionó accidentalmente la arteria coroidea anterior, por lo que se vio forzado a ligarla para prevenir la formación de un hematoma; sin embargo, este procedimiento ocasionó el inesperado control del temblor y la rigidez en el sitio contralateral a la lesión generada.68 La talamotomía fue la cirugía más utilizada en la década de 1950 a 1960, técnica empleada por Cooper en Estados Unidos y Hassler en Alemania, porque el tálamo ventrolateral era el sitio ideal para el control de los síntomas motores.69 Posterior a esta fecha, entre 1960 y 1970, con la introducción de la levodopa hubo una reducción acentuada en el empleo de la esterotaxia con fines quirúrgicos enfocados a la EP. A mediados de la década de 1980 renacieron las técnicas neuroquirúrgicas. Lauri Laitinen et al. reconocieron las limitaciones de los tratamientos farmacológicos y retomaron los procedimientos ablativos, particularmente la palidotomía posteroventral.70 En ese mismo periodo, específicamente en 1987, se identificó una función importante del núcleo subtalámico en la EP; múltiples equipos de científicos en distintas partes del mundo realizaron trabajos experimentales, los cuales fueron inicialmente realizados en modelos animales con parkinsonismo y posteriormente en humanos afectados con la enfermedad.71-73 Las observaciones de Bergman, Witchman y De Long, en relación con la respuesta con una lesión en el núcleo subtalámico, provee un arma importante para el desarrollo del que tal vez sea considerado uno de los más grandes descubrimientos de esa época: la estimulación cerebral profunda de alta frecuencia.74 El año de 1993 se considera un parteaguas en relación con la cirugía funcional: Alim-Louis Benabid reportó la implantación de electrodos en el núcleo subtalámico.75 Por lo anterior, se considera a 1987 como el año del nacimiento de la estimulación cerebral profunda y a 30 años de esta valiosa aportación, se dispone de diversos procedimientos de neurocirugía funcional y de estimulación cerebral profunda, los cuales han evolucionado a la par de las tecnologías contemporáneas, que también progresan incesantemente.
Aún queda mucho camino por recorrer, y es responsabilidad de los científicos y médicos de la actualidad continuar escribiendo la historia día a día con el fin de encontrar la cura, que nos ha eludido durante los últimos dos siglos.
Bibliografía
Keppel-Hesselink JM. Evolution of concepts and definitions of Parkinson’s disease since 1817. J Hist Neurosci. 1996; 5:200-207. 2. Hurwitz B. Urban observation and sentiment in James Parkinson’s essay on the shaking palsy (1817). Lit Med. 2014;32:74-104. 3. Parkinson J. Definition, history, illustrative cases. En: An essay on shaking palsy. Londres: Whittingham and Rowland; 1817. 4. León R. Historia de la levodopa; un tratamiento anunciado desde la antigüedad. AFT. 2007;5:205-207. 5. García P. Prehistoria de la enfermedad de Parkinson. Neurologia. 2004;19:735-737. 6. Jonker MA. Estimation of life expectancy in the Middle Ages. J R Statist Soc A. 2003;166:105-117. 7. Rosler R, Young P. La lección de anatomía del doctor Nicolaes Tulp: el comienzo de una utopía médica. Rev Med Chile. 2011;139:535-541. 8. Charcot JM. Lecture VII: On slow compression of the spinal cord. En: Lectures on the diseases of the nervous system. Londres: New Sydenham Society; 1881. 9. Charcot JM. De la paralysie agitante. En: Oeuvres complètes. Leçons sur les maladies du système nerveux. Tomo I. París: Bureaux du Progrès Médical; 1892. pp. 155-188. 10. Lewy FH. Zur pathologischen Anatomie der Paralysis agitans. Deutsche Zeitschrift für Nervenheilkunde. 1913;50:50-55. 11. Trétiakoff K. Contribution à l’étude de l’anatomie pathologique du locus niger de Soemmering avec quelques déductions relatives à la pathogénie des troubles du tonus musculaire et de la maladie de Parkinson. Francia: Universidad de París; 1919. 12. Bethlem J, Den-Hartog-Jager WA. The incidence and characteristics of Lewy bodies in idiopathic paralysis agitans (Parkinson’s disease). J Neurol Neurosurg Psychiatry. 1960;23:74-80. 13. Braak H, Del-Tredici K, Rüb U, De-Vos RA, Jansen-Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197-211. 14. Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979-980. 15. Angot E, Steiner JA, Hansen C, Li JY, Brundin P. Are synucleinopathies prion-like disorders? Lancet Neurol. 2010;9:1128-1138. 16. Hasegawa M, Nonaka T, Masuda-Suzukake M. Prion-like mechanisms and potential therapeutic targets in neurodegenerative disorders. Pharmacol Ther. 2017;172:22-33. 17. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dutra A, Pike B, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045-2047. 18. Nussbaum RL, Polymeropoulos MH. Genetics of Parkinson’s disease. Hum Mol Genet. 1997;6:1687-1691. 19. Jones AC, Yamamura Y, Almasy L, Bohlega S, Elibol B, Hubble J, et al. Autosomal recessive juvenile parkinsonism maps to 6q25.2-q27 in four ethnic groups: detailed genetic mapping of the linked region. Am J Hum Genet. 1998;63:80-87. 20. Bonifati V, Rizzu P, Van-Baren MJ, Schaap O, Breedveld GJ, Krieger E, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256-259. 21. Hatano Y, Li Y, Sato K, Asakawa S, Yamamura Y, Tomiyama H, et al. Novel PINK1 mutations in early-onset parkinsonism. Ann Neurol. 2004;56:424-427. 22. Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601-607. 23. Katzenschlager R, Evans A, Manson A, Patsalos PN, Ratnaraj N, Watt H, et al. Mucuna pruriens in Parkinson’s disease: a double blind clinical and pharmacological study. J Neurol Neurosurg Psychiatry. 2004; 75:1672-1677. 24. Guebila MB, Thiele I. Model-based dietary optimization for late-stage, levodopa-treated, Parkinson’s disease patients. NPJ Syst Biol Appl. 2016;2:16013. 25. Nagashima Y, Kondo T, Sakata M, Koh J, Ito H. Effects of soybean ingestion on pharmacokinetics of levodopa and motor symptoms of Parkinson’s disease: in relation to the effects of Mucuna pruriens. J Neurol Sci. 2016;361:229-234. 26. Ordenstein L. Étiologie, pronostic, thérapeutique. En: Sur la paralysie agitante et la sclérose plaque généralisée. Francia: A Delahaye, Libraire-Éditeur; 1868. 27. Charcot JM. Leçon 5. En: Oeuvres complètes de J.M. Leçons sur les maladies du système nerveux. Tomo I. Bureaux du Progrès Médical; París: 1892. 28. Gowers WR. Paralysis agitans. En: A system of medicine. Londres: Macmillan; 1899. 29. Gowers WR. Paralysis agitans. En: A manual of diseases of the nervous system. Londres: J. & A.; 1888. 30. Fahn S. The history of dopamine and levodopa in the treatment of Parkinson’s disease. Mov Disord. 2008;23:497-508. 31. Carlsson A, Waldeck B. A fluorimetric method for the determination of dopamine (3‐hydroxytyramine). Acta Physiol Scand. 1958;44:293-298. 32. Price KS, Farley IJ, Hornykiewicz O. Neurochemistry of Parkinson’s disease: relation between striatal and limbic dopamine. Adv Biochem Psychopharmacol. 1978;19:293-300. 33. Lloyd K, Davidson L, Hornykiewicz O. The neurochemistry of Parkinson’s disease: effect of L-dopa therapy. J Pharmacol Exp Ther. 1975;195:453-464. 34. Ehringer H, Hornykiewicz, O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system. Parkinsonism Relat Disord. 1960;4:53-57. 35. Montagu KA. Catechol compounds in rat tissues and in brains of different animals. Nature. 1957;180:244-245. 36. Blaschko H. The specific action of L-dopa decarboxylase. J Physiol. 1939;96:50-51. 37. Holtz P. Dopa decarboxylase. Naturwissenschaften. 1939;27:724-725. 38. Cotzias GC, Van-Woert MH, Schiffer LM. Aromatic amino acids and modification of parkinsonism. N Engl J Med. 1967;276:374-379. 39. Cotzias GC, Papavasiliou PS, Gellene R. Modification of Parkinsonism: chronic treatment with L-dopa. N Engl J Med. 1969;280:337-345. 40. Fahn S. Parkinson disease the effect of levodopa and the ELLDOPA trial. Earlier vs later L-dopa. Arch Neurol. 1999;56:529-535. 41. Doshay LJ, Constable K. Newer drugs in the treatment of parkinsonism. Neurology. 2001;57:4-10. 42. Corbin KB. Trihexyphenidyl; evaluation of the new agent in the treatment of Parkinsonism. J Am Med Assoc. 1949;141:377-382. 43. Bertol E, Fineschi V, Karch SB, Mari F, Riezzo I. Nymphaea cults in ancient Egypt and the New World: a lesson in empirical pharmacology. J R Soc Med. 2004;97:84-85. 44. Taba P, Lees A, Stern G. Erich Harnack (1852-1915) and short history of apomorphine. Eur Neurol. 2013;69:321-324. 45. Matthiessen A. Researches into the chemical constitution of the opium bases. Part I.- On the action of hydrochloric acid on morphia. Proc R Soc Lond. 1868;17:455-460. 46. Matthiessen A, Wright C. Apomorphia, a new base derived from morphine. Pharm J Tr. 1869;11-12. 47. Djamshidian A, Poewe W. Apomorphine and levodopa in Parkinson’s disease: two revolutionary drugs from the 1950’s. Parkinsonism Relat Disord. 2016;33:S9-S12. 48. Schwab RS, Amador LV, Lettvin JY. Apomorphine in Parkinson’s disease. Trans Am Neurol Assoc. 1951;56:251-253. 49. Ernst AM. Relation between the action of dopamine and apomorphine and their O-methylated derivatives upon the CNS. Psychopharmacologia. 1965;7:391-399. 50. Cotzias GC, Düby S, Ginos JZ, Steck A, Papavasiliou PS. Dopamine analogues for studies of parkinsonism. N Engl J Med. 1970;283:1289. 51. Cotzias GC, Papavasiliou PS, Tolosa ES, Mendez JS, Bell-Midura M. Treatment of Parkinson’s disease with aporphines: Possible role of growth hormone. N Engl J Med. 1976;294:567-572. 52. Stibe C, Lees A, Stern G. Subcutaneous infusion of apomorphine and lisuride in the treatment of parkinsonian on-off fluctuations. Lancet. 1987;1:871. 53. Lees AJ, Haddad S, Shaw KM, Kohout LJ, Stern GM. Bromocriptine in parkinsonism: a long-term study. Arch Neurol. 1978;35:503-505. 54. Pearce RK, Banerji T, Jenner P, Marsden CD. De novo administration of ropinirole and bromocriptine induced less dyskinesia than L‐dopa in the MPTP‐treated marmoset. Mov Disord. 1998;13:234-241. 55. Rascol O, Pathak A, Bagheri H, Montastruc JL. Dopaminagonists and fibrotic valvular heart disease: further considerations. Mov Disord. 2004;19:1524-1425. 56. Hobson DE, Pourcher E, Martin WR. Ropinirole and pramipexole, the new agonists. Can J Neurol Sci. 1999;26:S27-S33. 57. Metman LV, Gillespie M, Farmer C, Bibbiani F, Konitsiotis S, Morris M, et al. Continuous transdermal dopaminergic stimulation in advanced Parkinson’s disease. Clin Neuropharmacol. 2001;24:163-169. 58. Pfeiffer RF. Potential of transdermal drug delivery in Parkinson’s disease. Drugs Aging. 2002;19:561-570. 59. Jenner P, Könen-Bergmann, M, Schepers C, Haertter S. Pharmacokinetics of a once-daily extended-release formulation of pramipexole in healthy male volunteers: three studies. Clin Ther. 2009;31:2698-2711. 60. Auffret M, Le-Jeune F, Maurus A, Drapier S, Houvenaghel JF, Robert GH, et al. Apomorphine pump in advanced Parkinson’s disease: Effects on motor and nonmotor symptoms with brain metabolism correlations. J Neurol Sci. 2017;372:279-287. 61. Bar Am O, Amit T, Youdim MB. Contrasting neuroprotective and neurotoxic actions of respective metabolites of anti-Parkinson drugs rasagiline and selegiline. Neurosci Lett. 2004;355:169-172. 62. Schwab RS, England AC, Poskanzer DC, Young RR. Amantadine in the treatment of Parkinson’s disease. JAMA. 1969;208:1168-1170. 63. Foerster O, Penfield W. The structural basis of traumatic epilepsy and results of radical operation. Brain 1930; 53:99-119. 64. Meyers RA. Surgical procedure for the alleviation of postencephalitic tremor, with notes on the physiology of premotor fibers. Arch Neurol Psychiatry. 1940;44:455-459. Gaceta Médica de México. 2018;154 726 65. Bucy PC, Case TJ. Tremor, physiologic mechanism and abolition by surgical means. Archi Neurol. 1939;41:721-746. 66. Klemme RM. Surgical treatment of dystonia, paralysis agitans and athetosis. Arch Neurol Psychiatry. 1940;44:926. 67. Spiegel EA, Wycis HT, Marks M, Lee AJ. Stereotaxic apparatus for operations on the human brain. Science. 1947;106:349-350. 68. Cooper IS. Ligation of the anterior choroidal artery for involuntary movements; parkinsonism. Psychiatr Q. 1953;27:317-319. 69. Hassler R. The influence of stimulations and coagulations in the human thalamus on the tremor at rest and its physiologic mechanism. Proc Second Intl Congr Neuropath. 1:637-642. 70. Laitinen LV, Bergenheim AT, Hariz MI. Leksell’s posteroventral pallidotomy in the treatment of Parkinson’s disease. J Neurosurg. 1992;76:53-61. 71. Smith Y, Parent A. Neurons of the subthalamic nucleus in primates display glutamate but not GABA immunoreactivity. Brain Res. 1988;453:353-356. 72. Rodríguez-Oroz MC, Rodríguez M, Guridi J, Mewes K, Chockkman V, Vitek J, et al. The subthalamic nucleus in Parkinson’s disease: somatotopic organization and physiological characteristics. Brain. 2001; 124:1777-1790. 73. Aziz TZ, Peggs D, Sambrook MA, Crossman AR. Lesion of the subthalamic nucleus for the alleviation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism in the primate. Mov Disord. 1991;6:288-289. 74. Bergman H, Wichmann T, DeLong MR. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249: 1436-1438. 75. Pollak P, Benabid AL, Gross C, Gao DM, Laurent A, Benazzouz A, et al. Effects of the stimulation of the subthalamic n
Kenia Arredondo-Blanco,1 Rosalía Zerón-Martínez,1 Mayela Rodríguez-Violante1 y Amin Cervantes-Arriaga2 1 Clínica de Trastornos del Movimiento; 2 Laboratorio Clínico de Enfermedades Neurodegenerativas. Secretaría de Salud, Instituto Nacional de Neurología y Neurocirugía, Ciudad de México, México Gac Med Mex. 2018;154:719