EL CÁNCER ES MÁS QUE UNA SOLA ENFERMEDAD
Este magnifico estudio del cancer, esta copiado en parte y mi agradecimiento y felicitación a su autor.
Existen muchos tipos de cáncer. El cáncer se puede desarrollar en cualquier parte del cuerpo y se denomina según la parte del cuerpo corporal en el que se origina.
Existen dos categoría principales de cáncer:
Los cánceres hematológicos (cánceres de la sangre) son tipos de cáncer en los glóbulos sanguíneos, como es el caso con la leucemia, el linfoma y el mieloma múltiple.
Los cánceres de tumor sólido son aquellos tipos que se desarrollan en cualquier órgano, tejido o parte el cuerpo. Los tipos más comunes de cáncer con tumor sólido son el de seno, próstata, pulmón y el colorrectal.
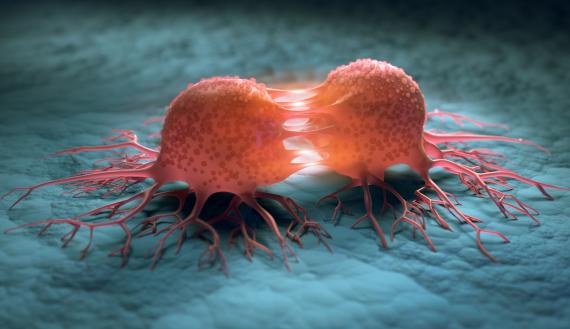
El cáncer es una enfermedad en la que las células se multiplican de forma descontrolada, invaden los tejidos circundantes y se extienden a distintas partes del organismo en un proceso llamado metástasis. La invasión y la metástasis son los rasgos clave que distinguen los tumores malignos —es decir, el cáncer propiamente dicho— de los benignos. El cáncer puede darse en principio en cualquier órgano del cuerpo y a cualquier edad (American Cancer Society 2008; Jemal et al. 2005). Sin embargo, por dura que resulte esta realidad, la idea de que es una enfermedad incurable debe contemplarse como un mito obsoleto. La mayoría de los cánceres pueden tratarse, muchos de ellos pueden controlarse con éxito y algunos curarse por completo. Los índices de curación para algunas clases de cáncer llegan al 95% de los casos, por encima de los de algunas enfermedades infecciosas y desórdenes metabólicos.
Fundamentalmente el cáncer es un problema genético. Surge a partir de mutaciones y otras alteraciones patológicas en el genoma de una célula, induciendo a ésta y a sus descendientes a un comportamiento anómalo (Volgestein y Kinzler 2004). Estas alteraciones pueden heredarse en el momento de la concepción y afectar a todas las células de un organismo, pero por lo común se adquieren por accidente en un pequeño número de células en un tejido en particular. En la mayoría de los tipos de cáncer la transformación de una célula normal en cancerígena requiere múltiples mutaciones que, unidas, desactivan los mecanismos clave del autocontrol celular. Este cúmulo de mutaciones puede tardar décadas en producirse, una de las razones por la cual la incidencia del cáncer aumenta con la edad.
El cáncer es también un problema de biología celular. Las alteraciones genéticas que dan lugar al cáncer actúan alterando el ciclo de vida normal y el comportamiento social de las células (Gupta y Massagué 2006; Hanahan y Weinberg 2000). Los genes cuya función normal sería favorecer el movimiento y la división de células pueden convertirse en cancerígenos si sufren alteraciones que incrementen dichas actividades. Por otra parte, los genes cuya función normal es la de limitar la división celular, retener a las células en su sitio, favorecer la diferenciación celular o eliminar células muertas o defectuosas, conducen al cáncer si no son activados como requieren. La identificación de células cancerígenas y de las funciones celulares que éstas controlan ocupa en la actualidad el primer plano de la investigación y del desarrollo de fármacos anticancerígenos.
La identificación de células cancerígenas y de sus funciones biológicas durante el último cuarto del siglo xx ha hecho posibles nuevas y mejores maneras de prevenir y tratar el cáncer. La mejora de los métodos de evaluación del riesgo de cáncer y de las campañas de prevención ha hecho disminuir la incidencia, así como la mortalidad, de ciertos tipos de cáncer. Procedimientos quirúrgicos menos invasivos, métodos de radioterapia más refinados y el perfeccionamiento de los medicamentos empleados en quimioterapia están contribuyendo al éxito creciente del tratamiento del cáncer convencional. Asimismo, una mejor comprensión de su biología y genética están posibilitando el desarrollo de nuevos y mejores medicamentos que tratan las células cancerígenas sin afectar a las sanas. Y aunque por ahora estos medicamentos aún llegan por goteo, vendrá el día en que este goteo se convertirá en inundación. La consecución de estos objetivos puede muy bien ser una de las grandes hazañas científicas del siglo xxi.
CRECIENTE INCIDENCIA DEL CÁNCER
El cáncer no es una enfermedad nueva. Los egipcios ya lo trataban quirúrgicamente alrededor del 600 a.C. (Karpozilos y Pavlidis 2004). Hacia el 400 a.C. Hipócrates distinguió entre tumores benignos y malignos; a los segundos los llamó «carcinomas», a partir de la voz griega carcinos, que significa «cangrejo», en referencia a la forma que observó en los tumores malignos en estado avanzado, y el sufijo -oma, que significa «inflamación». Pero aunque no es una enfermedad nueva, la incidencia del cáncer va en aumento. Las estadísticas más recientes cifran la mortalidad anual por cáncer en casi ocho millones de personas, alrededor del 13% del total de muertes (Organización Mundial de la Salud 2008). La Organización Mundial de la Salud también predice que en 2020 esta cifra habrá ascendido a 11,5 millones.
Los tumores resultan de la acumulación de múltiples mutaciones en las células afectadas. Esta acumulación puede llevar años (Volgestein y Kinzler 2004). Por esta razón el cáncer es relativamente raro en niños y adolescentes y en cambio aumenta con la edad. En los países desarrollados, el aumento de la esperanza de vida y de la población de mediana edad ocurrido en las últimas décadas ha contribuido a un crecimiento generalizado de la incidencia de cáncer. Dados los progresos conseguidos en el control de las enfermedades infecciosas que en la actualidad azotan a la población de los países en vías de desarrollo, cabe esperar que crezca también la incidencia de cáncer en ellos. Otros factores desencadenantes son la detección temprana de tumores en exámenes médicos rutinarios, factores relacionados con la dieta y los hábitos de vida y el impacto negativo del tabaco.
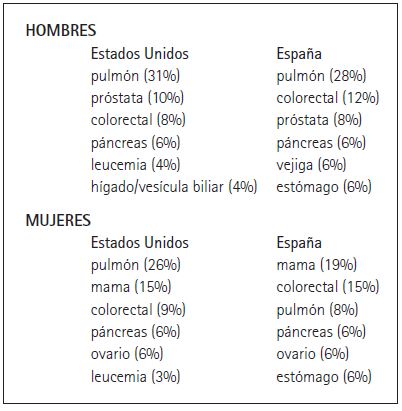
La incidencia del cáncer en general y la de determinados tipos de cáncer varía entre los países (Danaei et al. 2005; OMS 2008). Por ejemplo, los tipos de cáncer con mayores índices de mortalidad coinciden en Estados Unidos y España con una notable excepción: la mortalidad por cáncer de pulmón en las mujeres. Éste es el primer causante de muertes por cáncer entre la población masculina de ambos países. Sin embargo, al menos hasta hace poco tiempo, ocupaba el tercer puesto entre muertes por cáncer en mujeres en España (Cuadro 1). Esta diferencia se atribuye a que, comparadas con los hombres, las mujeres españolas empiezan a fumar más tarde en España que en Estados Unidos. Estudios epidemiológicos demuestran además una estrecha correlación entre tabaquismo y cáncer de pulmón, con una media de veinte años entre el inicio en el hábito y la aparición de la enfermedad.
CÁNCER Y CÁNCERES
El término «cáncer» agrupa a cientos de enfermedades diferentes. Tumores primarios que se originan en diferentes órganos y tejidos —por ejemplo, cáncer de mama, cáncer de pulmón o leucemia— son distintos entre sí en cuanto a apariencia, evolución, respuesta al tratamiento y mortalidad. Pero además, tumores originados en un mismo órgano pueden clasificarse en subclases que difieren mucho entre sí. Existen al menos cinco subclases de cáncer de mama, e incluso éstas podrían dividirse a su vez en diferentes variedades. Lo mismo puede decirse del cáncer en otros órganos. Estas diferencias requieren, pues, distintos tratamientos.
Los tumores también se clasifican de acuerdo al tipo de célula del que deriven. Los carcinomas son tumores malignos derivados de células epiteliales, tales como las que forman la capa superficial de la piel o epidermis, y en la mucosa digestiva o la estructura interna de órganos como la mama, la próstata, el hígado y el páncreas. Los sarcomas, por su parte, derivan de células de tejido conectivo como huesos, cartílagos y músculos. Los linfomas y las leucemias se originan en células precursoras; los melanomas en los melanocitos (células responsables de la pigmentación de la piel), y el glioblastoma, el neuroblastoma y el meduloblastoma, en células inmaduras del tejido neural. Los carcinomas son el tipo de cáncer más común en adultos, mientras que entre la población joven son más corrientes el neuroblastoma, el meduloblastoma y la leucemia.
Cuadro 1. Incidencia de cáncer en adultos en Estados Unidos (American Cancer Society, 2008) y España (Centro Nacional de Epidemiologia deEspaña). Las cifras entre paréntesis representan el porcentaje total de muertes por ese tipo concreto de cáncer.
Una tercera serie de parámetros en la clasificación de tumores se basa en su grado de invasión, lo que se conoce como estadios de la enfermedad, y su presentación histológica cuando se observan en el microscopio, llamada grado. Sin embargo, tumores de un mismo origen, tipo, grado y estadio pueden progresar y responder a la terapia de modos muy distintos dependiendo del paciente. Este hecho tiene un gran impacto en nuestra perspectiva del cáncer como enfermedad de la que aún sabemos muy poco. Por fortuna, esto está a punto de cambiar. La llegada de las tecnologías en genética molecular está haciendo posible una mejor clasificación de los tipos de cáncer basada en su origen específico, sus alteraciones moleculares, su riesgo de extenderse a otros órganos y su posible tratamiento.
CAUSAS DEL CÁNCER
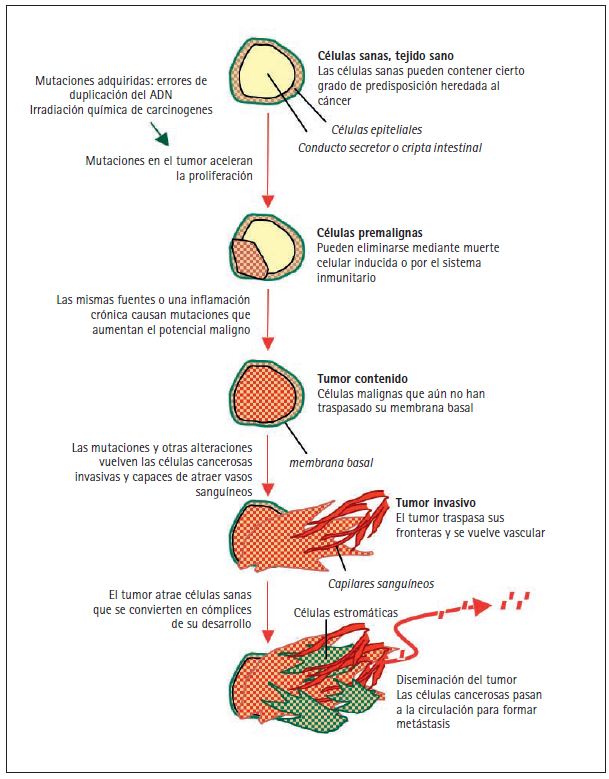
El cáncer se desarrolla como consecuencia de mutaciones y otras anormalidades que alteran los genes que controlan el comportamiento celular (Hanahan y Weinberg 2000; Volgestein y Kinzler 2004). Estas mutaciones pueden producirse por la acción de factores externos —cancerígenos químicos, agentes infecciosos o radioactivos— o por errores internos en la replicación y corrección del ADN de pequeños grupos de células a lo largo del tiempo (fig. 2). Las mutaciones cancerígenas también pueden ser hereditarias, en cuyo caso se encuentran presentes en las células desde el momento de nacer. Las investigaciones actuales sobre genética se centran en los procesos que causan estas alteraciones, en los tipos de genes que se ven afectados y en sus consecuencias biológicas.
Ejemplos comunes de cancerígenos químicos incluyen el tabaco, que causa cáncer de pulmón y de vejiga, y la exposición a fibras de asbesto, que causa mesotelioma (Danaei et al. 2005). La radiación ultravioleta del sol puede producir melanoma y otros tipos de cáncer de piel. Se cree que los agentes cancerígenos presentes en el tabaco y las radiaciones favorecen la formación de tumores al actuar como mutágenos directos. El tabaco y los asbestos también pueden causar inflamaciones crónicas que a su vez pueden favorecer el desarrollo de tumores.
Las infecciones virales son la segunda causa externa más importante de cáncer después del tabaco (Zur Hausen 1999). Los virus asociados al cáncer en seres humanos incluyen el virus del papiloma en cáncer cervical, los virus de las hepatitis B y C en el cáncer de hígado, el VIH en el sarcoma de Kaposi y el virus de Epstein-Barr en linfomas de células B (Boshoff y Weiss 2002; Parato et al. 2005; Roden et al. 2006; Woodman et al. 2007; Young y Rickinson 2004). Las infecciones virales favorecen la formación de tumores al incorporar el genoma del virus en el ADN de la célula huésped, lo que puede incrementar la actividad de genes vecinos, los cuales a su vez estimulan la división incontrolada de células. Las infecciones virales también pueden favorecer la formación de tumores causando inflamación crónica y estimulando la producción de células en los tejidos huésped. La degeneración del tejido hepático, o cirrosis, causada por el alcoholismo, está relacionada con el desarrollo de cáncer de hígado. La combinación de cirrosis y hepatitis viral constituye el principal factor de riesgo de cáncer de hígado, que es de los más comunes y con mayor índice de mortalidad. El ejemplo más claro son los cánceres gástricos relacionados con la inflamación crónica de la mucosa estomacal por infección de la bacteria Helicobacter pylori (Cheung et al. 2007; Wang et al. 2007).
Ciertos tipos de cáncer tienen un fuerte componente hereditario (Volgestein y Kinzler 2004). Mutaciones heredadas en los genes BRCA1 y BRCA2 crean un componente de alto riesgo de desarrollar cáncer de mama y de ovarios (Walsh y King 2007; Wang 2007; Welsch y King 2001). Es interesante que las mutaciones en genes BRCA son poco frecuentes en cáncer esporádico (aquel en el que las mutaciones genéticas no son hereditarias, sino espontáneas). Por contra, el p53, que por lo general muta en los casos de cáncer esporádico, también es el gen afectado en el síndrome hereditario de Li-Fraumeni, que incluye predisposición a sarcomas, cáncer de mama y tumores cerebrales (Vousden y Lane 2007). El retinoblastoma en niños se debe a una mutación de carácter hereditario en el gen retinoblastoma (RB), que también muta en muchos tipos de cáncer esporádico (Classon y Harlow). Una mutación heredada del gen APC también puede dar lugar a la aparición de cientos de pólipos en el colon conducentes a un desarrollo temprano de carcinoma de colon (Fodde et al. 2001). Otra forma hereditaria de predisposición al cáncer es la causada por mutaciones en uno de los varios genes (MLH1, MSH2, MSH6, PMS1, PMS2) dedicados a reparar errores de replicación en el ADN. Este trastorno hereditario (llamado HNPCC, cáncer de colon hereditario sin pólipos) incluye casos de cáncer de colon sin pólipos, cáncer uterino, gástrico y de ovarios dentro de una misma familia (De la Chapelle 2004). Mutaciones heredadas en el gen VHL1 producen una predisposición al cáncer de riñón (Kaelin 2005).
Las mutaciones hereditarias que tienen un fuerte efecto en el desarrollo del cáncer son raras entre los seres humanos y responsables sólo de una pequeña fracción de las estadísticas totales de cáncer. Por ejemplo, la mutación heredada del BRCA es responsable de menos de un 2% de los casos de cáncer de mama (Welsch y King 2001). En el extremo opuesto del espectro, ciertas variaciones genéticas pueden tener un impacto muy leve a escala individual en el riesgo de desarrollar cáncer, pero pueden ser prevalentes en los seres humanos. En determinados casos estos rasgos genéticos pueden combinarse para crear un riesgo significativo de cáncer. La visión predominante en la actualidad es que el cáncer surge a partir de complejas interacciones entre carcinógenos externos y el genoma individual. La identificación de estos determinantes genéticos en hoy día objeto de intensas investigaciones.
CÉLULAS SANAS Y CÉLULAS CANCEROSAS
La célula es la unidad básica de la vida. Aislada, sus actividades básicamente son resistir el entorno, incorporar nutrientes, replicar fielmente su genoma y dividirse. Sin embargo, las células que conforman los tejidos de un organismo complejo ya no son capaces de realizar estas tareas de forma autónoma. Células individuales evolucionaron y formaron colonias hace cientos de millones de años porque esta forma comunal de vida suponía una ventaja a la hora de enfrentarse a entornos hostiles. Pero también implicaba sacrificar cierto grado de libertad. Por ejemplo, ya no era posible para una célula dentro de una comunidad dividirse o desplazarse a voluntad. En nuestros tejidos altamente organizados, estas decisiones están sujetas a una complicada red de señales moleculares entre células. Esta forma de diálogo intercelular lleva desarrollándose y enriqueciéndose millones de años y una buena parte de nuestro genoma está enteramente dedicada a ella.
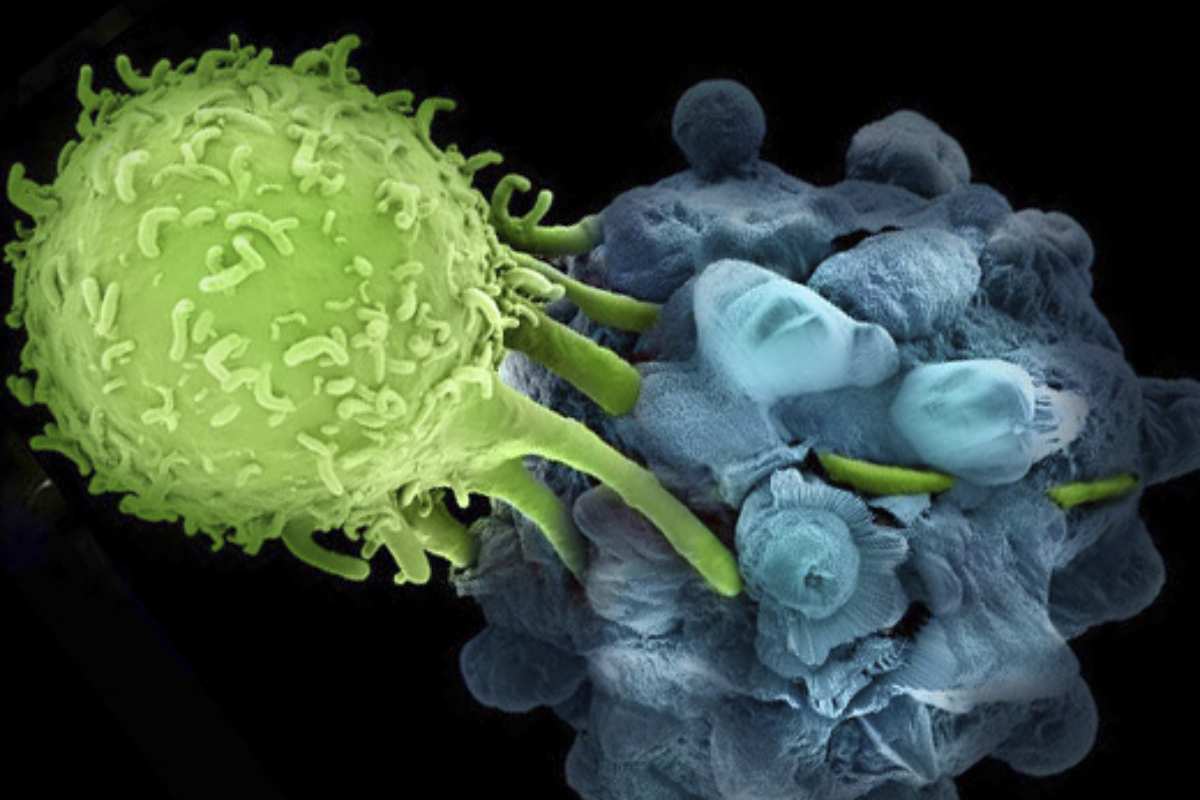
Las células se comunican unas con otras segregando moléculas, por lo general en forma de pequeñas proteínas llamadas hormonas, factores de crecimiento, citoquinas o quimiocinas. Estos factores contactan los receptores de proteínas en la superficie de las células de destino activando vías, que son secuencias de reacciones bioquímicas entre proteínas portadoras de señales dentro de la célula (Bierie y Moses 2006; Bild et al. 2006; Christofori 2006; Ciardello y Tortora 2008; Classon y Harlow 2002; Ferrara 2002; Fodde et al. 2001; Hanahan y Weinberg 2000; Karin 2006; Weinberg 2000; Malumbres y Barbacid 2007; Massagué 2004 y 2008; Olsson et al. 2006; Pouyssegur et al. 2006; Sweet-Cordero et al. 2005; Vousden y Lane 2007). El resultado final de este proceso son cambios positivos o negativos en la capacidad de la célula para desplazarse, metabolizar, crecer, dividirse, diferenciarse o morir. Otras proteínas del interior de la célula detectan la presencia de errores y alteraciones en el ADN y provocan, bien su reparación, bien su muerte. La pérdida de estas importantes funciones señalizadoras y de autocontrol deriva en la aparición de cáncer. Las células cancerosas desobedecen reglas esenciales de la vida en comunidad incrementando los estímulos proliferativos erróneos e ignorando las leyes de moderación. Su interacción con sus vecinas se vuelve abiertamente antisocial y escapan del control del sistema inmunitario. Con el tiempo rompen las barreras que encapsulan el tumor e inician un recorrido que diseminará las células cancerosas por el cuerpo, creando metástasis.
Las mutaciones causantes del cáncer afectan específicamente los genes encargados de ejercer estas funciones de control que tan importantes son. La acumulación progresiva de mutaciones convierte células normales en pre-malignas y, con el tiempo, en malignas (fig. 2). Estos cambios pueden observarse en el microscopio. Un proceso maligno puede empezar con la presencia de un número excesivo de células de apariencia normal, conocido como hiperplasia, y más específicamente con una acumulación desordenada de células de este tipo, llamada displasia. Cuando las células dejan de parecer normales la lesión se considera carcinoma in situ, en el cual las células anormales siguen estando dentro de los límites normales. Cuando las células del carcinoma invaden los tejidos adyacentes rompiendo su membrana o lámina basal, la lesión recibe el nombre de carcinoma invasivo. Cada una de estas etapas se acompaña de la progresiva acumulación de mutaciones que conducen al cáncer.
Las funciones específicas que deben ser perturbadas para que se generen células cancerígenas incluyen un aumento de autonomía en la emisión de señales inductoras de crecimiento; pérdida de sensibilidad a las señales inhibitorias de crecimiento; pérdida de la capacidad de muerte celular (llamada pérdida de apoptosis); aumento de la capacidad de replicar perpetuamente el ADN y aumento en la habilidad para escapar al control del sistema inmunitario (Hanahan y Weinberg 2000). Estos cambios son un requisito en todos los tipos de cáncer, incluidos los cánceres sanguíneos, la leucemia. Para formar un tumor, las células cancerosas procedentes de tejidos sólidos requieren además un aumento de la capacidad de resistir la hipoxia por medio de la inducción de nuevos capilares que alimenten el tumor (angiogénesis), así como el incremento de la capacidad de separarse e invadir los tejidos adyacentes (fig. 2). Para extender el tumor a distintos puntos dentro del organismo, las células cancerosas deben también adquirir la capacidad de pasar al sistema circulatorio, penetrar tejidos distantes y adaptarse al microentorno de dichos tejidos hasta terminar por apoderarse de ellos.
GENES DEL CÁNCER
Los genes del cáncer se dividen en dos grandes grupos. Aquellos cuyo exceso de actividad contribuye a la aparición de cáncer se denominan oncogenes (Hanahan y Weinberg 2000). Los genes codifican receptores de factores de crecimiento tales como el EGFR y el HER2, transductores de señales de crecimiento como RAS, RAF y P13K, factores de supervivencia celular como el BCL2 y otros. Las mutaciones que afectan a estos genes son activadoras o de «ganancia de función». Los genes cuya actividad normal previene la aparición del cáncer reciben el nombre de «supresores tumorales» y las mutaciones que los afectan en procesos cancerígenos son inactivadoras. Los supresores tumorales incluyen sensores del daño en el ADN como el p53, genes que reparan los daños en el ADN como los BRCA1 y BRCA2, inhibidores del ciclo de división celular como el RB, receptores y transductores de señales inhibidoras del crecimiento como el TGFBR y el SMAD4 y supresores de señales de crecimiento como el PTEN.
Las mutaciones que afectan a estos genes pueden ser puntuales, es decir, que afectan a un solo nucleótido del gen y a un solo aminoácido en el producto del gen. Las mutaciones puntuales pueden aumentar o reducir la actividad del producto del gen, y por tanto son una causa de activación oncogénica así como de la inactivación de genes supresores tumorales. Pequeñas pérdidas o inserciones también pueden causar activación oncogénica o inactivación de los supresores tumorales. Las mutaciones a gran escala incluyen pérdida o adquisición de una porción de cromosoma que resulta en la multiplicación de uno o más oncogenes, o una pérdida de genes supresores tumorales. Las translocaciones se producen cuando dos regiones cromosómicas diferenciadas se fusionan de forma irregular, a menudo en una localización determinada. Un ejemplo conocido de esto es el cromosoma Filadelfia o translocación de los cromosomas 9 y 22, que se da en la leucemia mieloide crónica y resulta en la producción de la proteína de fusión BCR-ABL (Melo y Barnes 2007). Ello causa activación oncogénica del gen ABL. Algunas mutaciones oncogénicas afectan no sólo a la región codificadora de proteínas de un oncogén sino a la región reguladora o «promotora» encargada de controlar el producto del gen. La inserción de un genoma viral cerca de la región promotora también puede llevar a la hiperactivación de un oncogén.
Además de las distintas clases de mutaciones que pueden alterar la estructura química de un gen normal convirtiéndolo en cancerígeno, los investigadores son cada vez más conscientes del impacto de las modificaciones epigenómicas. Éstas son modificaciones químicas del ADN y de las proteínas que lo rodean (Blasco 2007; Esteller 2007). Dichas modificaciones se conocen como cambios epigenéticos y tienen la capacidad de silenciar la expresión de un gen o de impedir que sea activado. La desregulación epigenética puede contribuir a la aparición de cáncer si no consigue silenciar la expresión de un gen o hacer que sea competente para su activación. La pérdida de metilación puede desembocar en una expresión aberrante de oncogenes. La metilación o acetilación de proteínas histonas que envuelven el ADN cromosómico también pueden sufrir alteraciones que favorezcan el cáncer. El fármaco experimental anti cancerígeno Vorinostat actúa restaurando la acetilación de las histonas y está actualmente en fase de prueba.
ECOLOGÍA DEL MICROENTORNO DE LOS TUMORES
Cada tejido tiene una estructura, unos límites y una vascularización característicos, además de un entorno extracelular de hormonas, nutrientes y metabolitos. Las células cancerosas que alteran estas condiciones están expuestas a fuentes de estrés medioambiental incluyendo falta de oxígeno (hipoxia) y nutrientes, acidez, estrés oxidativo y respuestas inflamatorias. Las células que sobreviven a estos agentes de desgaste se convierten en población dominante en el tumor en desarrollo. Este fenómeno se conoce como «selección clonal» (Nowell 1976). Los clones de células resultantes no son meros supervivientes sino oportunistas altamente efectivos que se benefician del microentorno del tumor.
Los tumores son más que un conglomerado de células cancerígenas, también incluyen células sanas que son atraídas y finalmente engullidas por el creciente tumor y pueden convertirse en cómplices de su desarrollo (Joyce 2005; Mueller y Fusenig 2004). El conjunto de tipos de células no cancerígenas presentes en un tumor se conoce como estroma y su importancia en el cáncer es cada vez más reconocida. Células endoteliales presentes en el tumor forman nuevos capilares sanguíneos que atraen nutrientes y oxígeno a la masa tumoral. Macrófagos y otras células inmunes e inflamatorias se congregan en el tumor en un intento por responder al daño infringido a los tejidos. Los macrófagos asociados a tumores producen factores de crecimiento y enzimas ECM degradadas que estimulan el crecimiento y la invasión de células cancerosas (Joyce 2005; Lewis y Pollard 2006). El tumor también recluta células de defensa ante el estrés del sistema circulatorio. Varios tipos de células de la sangre son atraídas por señales que emanan del tumor y proliferan en respuesta a ellas. Los factores derivados del estroma pueden a su vez estimular a las células cancerosas a liberar señales que refuercen su capacidad de producir metástasis. Por ejemplo, la citoquina derivada de estroma factor de crecimiento transformante tipo b (TGF-b) puede inducir a las células de cáncer de mama a liberar angiopoyetina-like 4, que refuerza la capacidad de estas células de invadir los pulmones antes de escapar del tumor primario (Padua et al. 2008). Así, el estroma de un tumor puede proporcionar ventajas metastásicas a las células cancerosas.
METÁSTASIS: LA EXTENSIÓN LETAL DE LOS TUMORES
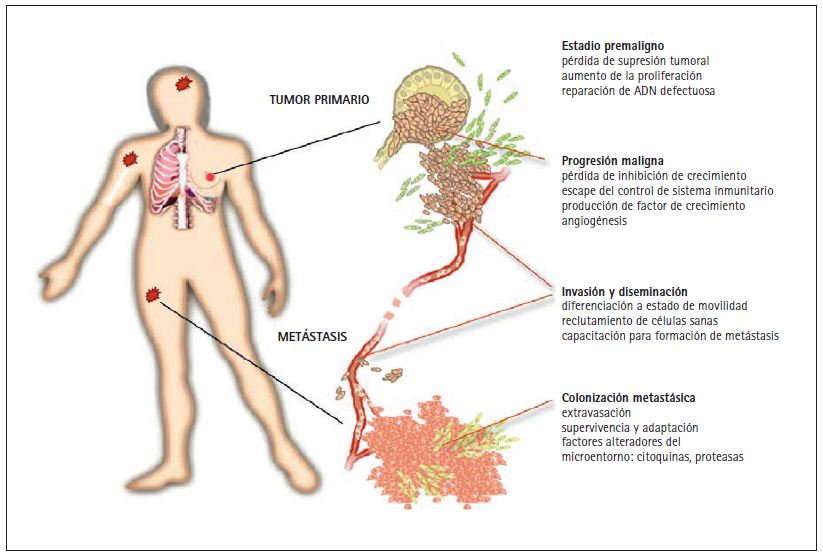
Los tumores agresivos pueden enviar millones de células cancerosas al sistema circulatorio antes de ser detectados y extirpados quirúrgicamente. La metástasis es el proceso por el cual estas células cancerosas diseminadas invaden distintos órganos y terminan por causar disfunción en los mismos y, en última instancia, la muerte (fig. 1). Las metástasis pueden detectarse coincidiendo con el diagnóstico inicial o meses o años más tarde, cuando se produce una recidiva. Las células cancerosas diseminadas pueden permanecer en estado letárgico en distintos órganos durante largos periodos de tiempo, hasta que, por causas que se desconocen, se reactivan y empiezan a formar metástasis de crecimiento agresivo.
Figura 1. Fases de un tumor sólido. Los tumores sólidos tales como carcinomas de pulmón, colon, mama o próstata empiezan en las células epiteliales que se alinean en la superficie de los bronquios, de la mucosa intestinal o de los alveolos o secreción mucosa de las mamas y la próstata. Las mutaciones que aumentan la capacidad de progresión de estas células generan pequeñas masas pre-malignas de tejido. Estas lesiones pre-cancerosas pueden progresar y convertirse en tumores malignos si experimentan nuevas mutaciones que las liberen de los controles inhibidores de crecimiento, de la protección del sistema inmunitario y las doten de capacidad de invadir los tejidos adyacentes y de atraer capilares sanguíneos (angiogénesis). Una nueva conversión de los tumores malignos conduce a la formación de células cancerosas altamente invasivas y de gran movilidad y al reclutamiento de células sanas que ayudan a diseminar el tumor. Estos cambios preparan el camino para que las células cancerosas penetren el sistema linfático y la circulación sanguínea y lleguen a todas las partes del cuerpo. Algunas células cancerosas diseminadas pueden tener la capacidad de salirse de la circulación (extravasación) traspasando las paredes de los capilares sanguíneos. Una vez penetran órganos distantes como la médula ósea, los pulmones, el hígado o el cerebro, las células cancerosas pueden sobrevivir, adaptarse y finalmente conquistar estos nuevos entornos, dando lugar a la formación de metástasis letales.
La administración de quimioterapia a pacientes de cáncer después de haberles extirpado un tumor primario tiene por objeto eliminar todas las células tumorales residuales y evitar la formación de metástasis. Sin embargo, el fracaso de las terapias existentes en la actualidad de curar metástasis es responsable del 90% de las muertes por cáncer. Si no fuera por las metástasis, el cáncer supondría únicamente una pequeña fracción del problema que es hoy en día. La comprensión de los múltiples procesos moleculares que participan en la formación de metástasis puede con el tiempo conducir a formas más efectivas de tratar esta enfermedad.
Los recientes avances tecnológicos a la hora de identificar y rastrear las metástasis han ayudado a perfilar los múltiples procesos que llevan a las células cancerosas de un tumor primario a alcanzar y colonizar órganos distantes (Fidler 2003; Gupta y Massagué 2006; Weinberg 2007) (fig. 2). Las células del carcinoma deben primero traspasar la membrana basal del tejido en que se encuentra el tumor. La membrana basal separa el epitelio de la célula en el que se ha originado el tumor del tejido subyacente. Las membranas basales también envuelven los vasos sanguíneos. Para traspasar la membrana y extenderse por el tejido adyacente las células cancerosas deben adquirir la capacidad de separarse de su lugar de origen, adoptar un comportamiento migratorio y liberar enzimas proteolíticas que degraden el armazón proteínico de la membrana basal y de la matriz extracelular.
Figura 2. Origen de las mutaciones cancerosas. La ilustración representa la sección de un conducto secretor o cripta intestinal con una capa de células epiteliales rodeadas de una membrana basal recubriendo la cavidad. La herencia genética de cada individuo contiene un nivel determinado —alto o bajo— de predisposición
a diferentes tipos de cáncer. Las variaciones genéticas de predisposición al cáncer que contienen un riesgo bajo de desarrollar un tipo determinado de cáncer son probablemente comunes en la población humana, y la mayoría están aún por descubrir. Las mutaciones heredadas que contienen un riesgo alto de desarrollar cáncer (por ejemplo, mutaciones BRCA1 y BRCA2 en cáncer de mama y ovario, mutaciones RB en retinoblastoma y mutaciones AP en carcinoma colorrectal) son poco frecuentes en la población. Estas predisposiciones intrínsecas al cáncer están presentes en todas las células del cuerpo. Sin embargo, el inicio de la formación de un tumor requiere, en todos los casos, que se produzcan más mutaciones. El origen de las mutaciones cancerosas puede ser interno, como errores no reparados en la replicación de ADN que las células sanas realizan por sí solas, o externo, como los carcinógenos químicos presentes en el tabaco o las radiaciones ultravioleta del sol. Estas mutaciones adquiridas aceleran la proliferación celular y conducen a la formación de lesiones pre-malignas, como pólipos intestinales o hiperplasias de tejido mamario. La mayoría de estas lesiones no progresan y son eliminadas por la autodestrucción celular o por el sistema inmunitario. Sin embargo, algunas lesiones pre-malignas pueden progresar hasta convertirse en un carcinoma in situ por la acumulación de mutaciones adicionales de origen
externo o causadas por la inestabilidad genómica de las células pre-cancerosas. Esta progresión también puede producirse por síndromes de infl amación crónica desencadenados por un sistema inmunitario defi citario (por ejemplo, la colitis ulcerosa), un elemento irritativo externo (el tabaco en los pulmones) o un agente infeccioso (por ejemplo el virus de la hepatitis en el hígado, la bacteria Heliobacter pilory en el estómago). Un tumor se convierte en carcinoma invasivo cuando rompe la membrana basal que lo rodea y atrae a los vasos capilares para proveerse de oxígeno y nutrientes. Las alteraciones epigenómicas en las células cancerosas y el estrés en los tejidos adyacentes pueden causar la liberación de factores que reclutan células sanas, que terminan colaborando en la progresión del tumor. Llegada esta fase, las células cancerosas tienen acceso a la circulación y pueden diseminarse por el organismo. Algunas de estas células diseminadas pueden también reproducir el tumor en órganos distantes, causando la formación de metástasis.
Una vez las células cancerosas han formado una pequeña masa tumoral y creado condiciones de hipoxia, responden a ésta mediante la secreción de citoquinas, las cuales estimulan la formación de nuevos capilares que las proveen del oxígeno y los nutrientes necesarios para el crecimiento del tumor. Como resultado de estos factores de permeabilidad derivados del tumor, estos capilares nuevos son porosos y permiten que las células cancerosas escapen y entre en la circulación sanguínea. Los vasos linfáticos que filtran fluidos del tumor al tejido adyacente proporcionan una nueva ruta para la diseminación de células cancerosas. Los nódulos linfáticos a menudo atrapan células cancerosas en circulación y documentan su expansión, por eso el estado de los nódulos linfáticos constituye un importante indicador de prognosis en el diagnóstico inicial. Sin embargo, la diseminación de células cancerígenas a órganos distantes tales como los pulmones, el cerebro, los huesos y el hígado se produce principalmente a través de la circulación sanguínea. En ella las células cancerosas se asocian unas con otras y con células sanguíneas para formar embolias que pueden contribuir a resistir la tensión mecánica o a escapar de la vigilancia del sistema inmunitario. 
Tumor de Hipofisis
Una vez las células cancerígenas se alojan en los capilares de órganos distantes deben atravesar las paredes capilares para acceder a la parénquima del órgano (fig. 3). La extravasación, como se conoce este proceso, depende de la capacidad de las células cancerosas de alterar los estrechos contactos existentes entre las células endoteliales de la pared capilar y de la membrana basal. El microentorno del órgano infiltrado es en gran medida hostil a las células cancerosas extravasadas, muchas de las cuales mueren. Aquellas que sobreviven forman micrometástasis que deben adaptarse al nuevo entorno y modificar las células residentes en él para reiniciar el proceso de crecimiento del tumor y formar colonias de metástasis agresivas. Este proceso puede llevar meses, años e incluso décadas. Tan sólo una pequeña fracción de las células cancerosas liberadas por un tumor cumple todos estos requisitos, pero las pocas que lo hacen bastan para que se formen metástasis letales.
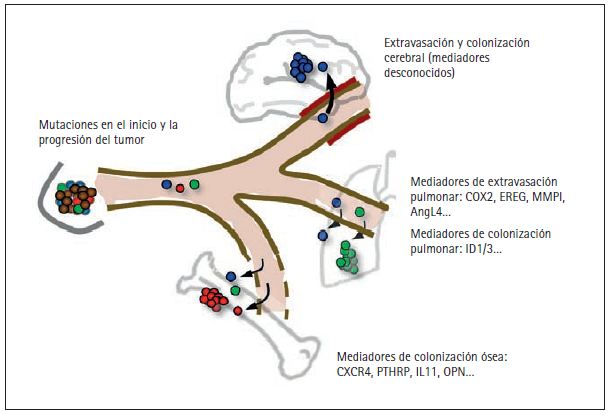
Figura 3. Mediadores en la formación de metástasis distantes en el cáncer de mama. He elegido para mi estudio las metástasis producidas por cáncer de mama debido a la alta incidencia de este tipo de tumores, la disponibilidad de material clínico y el número de órganos que pueden verse afectados.
Los tumores mamarios pueden liberar células en la circulación sanguínea tan pronto como se vuelven localmente invasivos, experimentando mutaciones que favorecen el origen y la progresión del tumor (fi g. 2). Las células diseminadas que sobreviven al estrés físico de la circulación requieren de nuevas funciones para penetrar tejidos distantes. El paso a dichos tejidos atravesando las paredes de los capilares sanguíneos o extravasación es relativamente permisivo en la médula ósea (y en el hígado, aunque no se ilustra aquí), porque los capilares de estos tejidos poseen ventanas naturales que permiten la entrada y salida constantes de células sanguíneas. Sin embargo, una vez penetran la médula ósea las células cancerosas deben ser capaces de sobrevivir e interactuar de forma productiva con este microentorno. El hecho de que las metástasis causadas por cáncer de mama pueden tardar años e incluso décadas en aparecer sugiere que las células cancerosas diseminadas llegaron originalmente a este órgano sin estar preparadas y tuvieron que desarrollar poco a poco las capacidades necesarias para expandirse en forma de agresivas colonias. Los genes de los que hacen uso las células de mama cancerosas para sobrevivir en la médula ósea incluyen el regulador de la quimiotaxis CXCR4, la proteína relacionada con la hormona paratiroidea estimuladora de osteoclastos PTHrP, el interleuquin-11 (IL11), el osteopontín (OPN) y otros genes. Al contrario de lo que ocurre con los capilares de la médula ósea, los capilares de otros órganos como los pulmones y en especial el cerebro tienen paredes resistentes que restringen el paso de las células en circulación. Para penetrar en estos órganos las células cancerosas deben por tanto transportar determinados genes activados. Entre ellos están el ligando epiregulina EGFR (EREG), la enzima cicloxigenasa-2 inhibidora de prostaglandina (COX2), la enzima metaloproteinasa-1 degradadora de colágeno (MMP1) y el factor alterador del endotelio angipoyetina-like 4 (AngL4). Se sospecha que algunos de estos genes también contribuyen a la penetración de células de mama cancerosas en el cerebro. Los genes que hacen posible la colonización de los pulmones y el cerebro son en gran medida desconocidos y sujeto de activas investigaciones. El ID1 y el ID3 han sido identificados recientemente como mediadores en la reiniciación de tumores por la penetración de células de mama cancerosas en el cerebro. Así, la expresión del ID1/3 es una propiedad de las células propagadoras de tumores, también conocidas como células madre cancerosas.
COMPONENTES DE LA METÁSTASIS. HETEROGENEIDAD GENÉTICA
La formación de metástasis tiene mucho de proceso evolutivo darwiniano: de una población de células cancerosas sólo las más fuertes sobreviven a las condiciones de su entorno. La evolución requiere de la presencia de heterogeneidad genética en una población en la que los individuos más fuertes pueden ser seleccionados para sobrevivir bajo determinadas presiones ambientales. En los tumores, dicha heterogeneidad viene garantizada por la inestabilidad genómica inherente a las células cancerosas, y aumenta la probabilidad de que algunas de las células de un tumor adquieran competencia metastásica. La integridad del ADN puede verse comprometida por aberraciones en la progresión del ciclo celular, crisis teloméricas, desactivación de los genes reparadores de ADN y alteración de los mecanismos de control epigenético. Por ejemplo, la mitad de los cánceres en seres humanos sufren pérdida del supresor tumoral p53, una proteína interna que responde a los daños en el ADN causando la eliminación de la célula dañada. La pérdida de p53 permite que las células cancerosas con alteraciones en el ADN sobrevivan y experimenten sucesivas mutaciones (Halazonetis et al. 2008). Mutaciones heredadas en determinados genes reparadores de ADN están asociadas a un riesgo mayor de desarrollar cáncer. Es el caso del síndrome de cáncer colorrectal sin poliposis hereditario (Rustgi 2007) y de cánceres de mama causados por mutaciones en el BRCA1 o el BRCA2 (Marin et al. 2008).
CÉLULAS MADRE CANCEROSAS
Las células cancerosas pueden diseminarse a partir de un tumor en estadios muy tempranos del mismo. Se han detectado células cancerosas en la médula ósea de pacientes con pequeños tumores de mama (Klein et al. 2002; Schmidt-Kittler et al. 2003). Ello no significa necesariamente que las primeras células migratorias sean las que progresan hasta convertirse en metástasis, pero sí indica que la diseminación no es propiedad exclusiva de tumores grandes y en estado avanzado.
Una vez las células diseminadas alcanzan órganos distantes pueden permanecer en estado letárgico e incluso morir. El estado letárgico puede durar años, incluso décadas hasta que las células cancerosas diseminadas inicien un crecimiento agresivo, como en el caso del cáncer de mama. Células cancerosas diseminadas encontradas en la médula ósea de mujeres o de ratones transgénicos con cánceres en estadios tempranos pueden activarse por trasplantes de médula ósea y causar tumores letales (Huseman et al. 2008).
La diseminación también puede darse en tumores metastásicos, que a su vez generan nuevas metástasis. Es posible que las células tumorales en circulación regresen a los mismos tumores de los que salieron. De acuerdo con esta hipótesis los tumores pueden alimentarse constantemente de su progenie más agresiva, proporcionando un mecanismo que aúna capacidad metastásica con crecimiento tumoral (Norton y Massagué 2006). Esto explicaría la persistente correlación entre metástasis y tamaño del tumor (Minn et al. 2007). El ritmo y los mecanismos de la diseminación de células cancerosas son objeto de gran atención en la investigación científica actual sobre cáncer.
DIFERENTES «SEMILLAS» PARA «SUELOS» DISTINTOS
Los huesos, los pulmones y el cerebro son las localizaciones más frecuentes de metástasis (fig. 3). Sin embargo, los diferentes tipos de cáncer son proclives a extenderse a diferentes órganos (Billingsley et al. 1999; Gavrilovi y Posner 2005; Hess et al. 2006; Leiter et al. 2004). La hipótesis sobre compatibilidad entre células cancerosas diseminadas (la «semilla») y determinados órganos distantes («suelos») ya la formuló en el siglo xix Stephen Paget (Paget 1889). Así por ejemplo, el cáncer de mama puede extenderse a estos cuatro órganos, aunque los huesos y los pulmones son los más frecuentemente afectados. Las metástasis de cáncer de pulmón se localizan sobre todo en el cerebro, los huesos y el otro pulmón. Por contra, las metástasis de cáncer de próstata se dan sobre todo en los huesos y, en mucha menor medida, en los pulmones. Es más, aunque estos tres tumores se extienden a los huesos, en ellos forman lesiones muy distintas: las metástasis de hueso por cáncer de mama y de pulmón son osteolíticas, es decir, que se disuelven en la matriz ósea causando fracturas. En cambio la metástasis de cáncer de próstata es osteoblástica, es decir, que genera tejido óseo anormal que llena la cavidad medular. La preferencia de un tumor en un órgano por hacer metástasis dentro de ese mismo órgano también varía: los tumores en un pulmón hacen fácilmente metástasis en el otro, mientras que esto rara vez ocurre en el cáncer de mama.
HACIA UNA COMPRENSIÓN DE LAS METÁSTASIS
Los progresos científicos que han acompañado la llegada del siglo xxi hacen posible una nueva visión de las metástasis basada en una mejor comprensión de sus fundamentos genéticos, moleculares y biológicos. Este conocimiento se acumula a gran velocidad a partir de la identificación de genes cuya actividad aberrante favorece la aparición de células metastásicas. Gracias a estos avances la metástasis está pasando de ser un oscuro objeto de estudio a un problema susceptible de analizarse racionalmente, diseccionarse y, en última instancia, resolverse.
UN MODELO INTEGRADO DE METÁSTASIS
Las primeras teorías sobre las metástasis proponían modelos antagónicos de predeterminación genética de una masa tumoral a metastatizar frente a la progresión del tumor resultante en la aparición de células anormales susceptibles de formar metástasis (Volgestein et al. 1998). Con la secuenciación del genoma humano se han desarrollado potentes tecnologías de microarray (microconfiguración) que permiten a los investigadores determinar el estado de actividad de cada gen de una pequeña muestra de tejido. Empleando dichas técnicas ha sido posible identificar patrones de actividad génica o «perfiles de expresión génica», capaces de indicar las probabilidades de que un tumor particular cause metástasis. Si una muestra extraída de un tumor primario revela la presencia de un perfil de expresión génica pro metastásico, ello indicaría que una proporción sustancial de las células de dicho tumor están expresando genes de este tipo y por tanto reúnen las condiciones —es decir, son competentes— para que se formen metástasis. Ello apoyaría la teoría sobre la predeterminación de las metástasis. Sin embargo, esta supuesta competencia de los genes puede ser incompleta. Han de producirse alteraciones adicionales antes de que las células cancerosas estén completamente equipadas para invadir y colonizar un tejido distante. La adquisición de todas las condiciones necesarias para que se formen metástasis puede darse de forma masiva en la población de un tumor, como es el caso de tumores que metastatizan rápidamente en múltiples órganos, o bien puede producirse lentamente en una minoría de células especialmente predispuestas, dando lugar a metástasis en uno u otro órgano, años o incluso décadas después de haber salido del tumor primario. Este último caso confirmaría la teoría de la progresión del tumor como un requisito necesario para la formación de metástasis.
Progresos recientes en el estudio de las metástasis han proporcionado pruebas experimentales y clínicas para ambos modelos, el de predeterminación y el de progresión, con el resultado de la formulación de un tercer modelo que integra los dos. Las células cancerosas de un tumor con prognosis desfavorable pueden contener genes activados que las doten de algunas, aunque no todas, las funciones requeridas para la formación de metástasis distantes. A estos genes los llamamos genes de progresión metastásica, porque permiten directamente a la población de células cancerosas adquirir la competencia necesaria para que se dé el comportamiento metastásico. Los genes de progresión metastásica son necesarios pero no bastan para crear metástasis, porque la mayoría de las células cancerosas que expresan dichos genes son todavía incapaces de formar tumores metastásicos. Esto implica la existencia de un conjunto complementario de genes metastásicos que proporcionan funciones adicionales de supervivencia y adaptación en un órgano determinado. A estos genes los llamamos de virulencia metastásica.
GENES DE PROGRESIÓN METASTÁSICA
Recientes trabajos realizados en nuestro laboratorio han identificado un conjunto de dieciocho genes que utilizan las células de mama cancerosas para proliferar tanto en el tumor primario como en los pulmones (fig. 3). Este conjunto, al que llamamos «perfil de expresión génica de metástasis de pulmón» o LMS incluye EREG, COX-2 y MMP1, los cuales cooperan en al remodelación de nuevos capilares sanguíneos en tumores mamarios y de los ya existentes en los pulmones cuando las células cancerosas favorecen la unión de capilares porosos que facilitan la salida de células cancerosas; en el pulmón estos mismos productos facilitan el paso de células cancerosas en circulación en la parénquima (Gupta et al. 2007). Otro ejemplo es el gen que codifica el ID1, que inhibe la diferenciación celular y estabiliza la capacidad de las células cancerosas de propagar el tumor. En modelos experimentales el ID1 es importante para el crecimiento de tumores de mama y para la reiniciación del crecimiento del tumor una vez las células cancerosas han alcanzado los pulmones. Así, los genes de progresión metastásica pueden cumplir los requisitos en cuanto a tejido del microentorno en un órgano particular para influir en la progresión del tumor primario. Pacientes de cáncer de mama con tumores primarios positivos en LMS tienen un riesgo mayor de desarrollar metástasis de pulmón, pero no de hueso o de otros órganos.
No todos los genes metastásicos que se expresan en tumores primarios proporcionan una ventaja selectiva en dichos tumores. Por ejemplo, la producción del factor transformador del crecimiento beta (TGFbeta) en el estroma de tumores primarios de mama estimula la expresión de más de cien genes en las células cancerosas mamarias del mismo tumor. Entre ellos está el gen que contiene el factor secretado ANGPTL4. A diferencia de EGFR, el COX2, el MMP1 o el ID1, la producción de ANGPTL4 no parece proporcionar ventaja ninguna a las células cancerosas en los tumores primarios, sino que únicamente refleja la presencia de TGFbeta en el tumor. Sin embargo, cuando las células cancerosas estimuladas llegan a los capilares del pulmón, el ANGPTL4 que secretan causa la rotura de la pared capilar y permite que el cáncer penetre en tejido (Padua et al. 2008).
CONTRIBUCIONES ESPECÍFICAS DE LOS GENES ASOCIADOS A LA VIRULENCIA METASTÁSICA
Cuando las células cancerosas alcanzan órganos distantes por lo general se enfrentan a un microentorno no permisivo. Para formar una colonia de metástasis el cáncer debe ser capaz de resistir y explotar su microentorno. Claro ejemplo de ello son las metástasis osteolíticas de hueso causadas por el cáncer de mama. Las células cancerosas en circulación que penetran la médula ósea deben encontrar la forma de sobrevivir en el entorno hormonal y estrómico de este tejido, así como la manera de activar la movilización y acción de los osteoclastos que hacen posible la destrucción de los huesos. Las células de mama cancerosas que forman metástasis de hueso muestran niveles elevados de CXCR4. Esta proteína de membrana actúa como receptora para el factor de supervivencia celular CXCL12, que se produce en grandes cantidades en el estroma de la médula ósea (Wang et al. 2006). Por tanto las células cancerosas que expresan niveles altos de CXCR4 obtienen una ventaja específica de la presencia de CXCL12 en la médula ósea.
En modelos experimentales que emplean ratones, las células de mama cancerosas que colonizan preferentemente los huesos muestran no sólo una elevada expresión del receptor de supervivencia celular CXCR4 sino también una alta producción de los factores PTHrP (péptido relacionado con la paratiroidea), TNF-alfa, IL-1, IL-6 e IL-11 (Kang et al. 2003). Cuando son secretados por células óseas metastásicas, estos factores estimulan a los osteblastos para que liberen RANKL, el cual activa la diferenciación osteoblástica. Los osteoblastos disuelven el hueso y liberan factores de crecimiento como el IGF-1 o factor de crecimiento insulínico, que favorece la supervivencia de las células cancerosas, y TGFbeta, que estimula las células cancerosas para que continúen segregando PTHrP. El resultado final de este proceso es un ciclo vicioso de interacciones entre osteoclastos y células cancerosas que aceleran la acción destructiva de las metástasis óseas.
La búsqueda actual de genes y funciones que intervienen en la formación de metástasis en forma de tumores de otra clase o en otros órganos comienza a dar resultados. Las células de cáncer de próstata segregan factores como el Wnt y proteínas óseas morfogenéticas (BMP) que estimulan la acumulación de osteoblastos. Como resultado de ellos el cáncer de próstata da lugar a metástasis osteoblásticas (producción anormal de hueso), en contraste con las metástasis producidas por el cáncer de mama, que causan la destrucción de los huesos. Comparadas con las metástasis en hueso y pulmón es muy poco lo que sabe acerca de los genes que usan las células cancerosas para colonizar el hígado o el cerebro. Sin embargo este tema es objeto de intensas investigaciones que a buen seguro terminarán por dar fruto en un futuro no lejano.
FRONTERAS EN LA PREVENCIÓN, DIAGNÓSTICO Y TRATAMIENTO DEL CÁNCER
Las campañas de prevención del cáncer enfocadas a reducir los comportamientos de riesgo (consumo excesivo de tabaco y alcohol, exceso de exposición al sol y otros), así como los exámenes médicos rutinarios en los que se detectan tumores en estadios tempranos son cruciales para reducir la incidencia y la mortalidad de esta enfermedad. El diagnóstico precoz hace posible intervenir el tumor antes de que éste se disemine, curando al paciente o al menos alargando su esperanza de vida. Se ha avanzado mucho en la detección de cáncer de mama por medio de mamografías, de cáncer colorrectal con colonoscopias, de cáncer de útero mediante citología y de cáncer de próstata con exámenes rectales y análisis de sangre que miden los niveles del PSA, o antígeno prostático específico. La vacunación preventiva contra el virus del papiloma humano de transmisión sexual tiene como fin reducir la incidencia del cáncer cervical. Se realizan exámenes genéticos para detectar ciertas mutaciones relacionadas con el cáncer en el BRCA1 y el BRCA2 (factores que predisponen a los cánceres de mama y ovarios) y para genes reparadores de ADN (que indican predisposición al cáncer de colon y otros) en individuos considerados de alto riesgo, con historial familiar de dichas enfermedades. Los portadores de estas mutaciones pueden así ser vigilados de cerca e incluso optar por cirugía profiláctica (mastectomías, extirpación de ovarios y de colon) para así reducir los riesgos de desarrollar el tumor.
La nueva estrategia terapéutica, que implica a patólogos, cirujanos, oncólogos o radioterapeutas, consiste en administrar la quimio-inmunoterapia antes de cirugía en pacientes en estadios precoces. »
Se trata de un cambio en el abordaje y la estrategia terapéutica. Hemos encontrado una mejora significativa que puede encaminarnos a la cura de un gran número de estos pacientes después de décadas sin avances»
Avances recientes están mejorando los enfoques clásicos en el tratamiento del cáncer (cirugía, quimioterapia, radioterapia) y los nuevos están basados en terapia dirigida e inmunoterapia. Los métodos quirúrgicos han ganado en precisión y son menos invasivos. La extirpación quirúrgica de un tumor que no se ha extendido puede curar el cáncer de forma efectiva. Sin embargo, la propensión de células cancerosas a invadir los tejidos adyacentes y extenderse a localizaciones distantes limita considerablemente la eficacia de la cirugía. Incluso tumores pequeños y localizados tienen potencial metastásico. Por tanto la cirugía se complementa a menudo con otras terapias. La radioterapia se basa en el uso de radiaciones ionizantes (rayos X) para encoger los tumores antes de la cirugía y eliminar células diseminadas localmente. La radiación puede dañar los tejidos sanos, por lo que sólo se aplica en áreas limitadas del cuerpo. La radioterapia destruye células causando extensos daños a su ADN. La mayoría de las células sanas puede recuperarse después de la radioterapia, lo que ofrece una ventana de posibilidad de que el tratamiento funcione.
Este enfoque de tratamiento, también se consigue una mayor la tasa de respuesta objetiva global (reducción o desaparición del tumor), ya que con quimio-innmunoterpia respondieron el 75,4 por ciento de los pacientes, en comparación con el 48,2 por ciento en el grupo de control. Además, permitiría elevar el número de pacientes que finalmente pueden ser operables: el 93 por ciento de los pacientes del grupo de terapia combinada se sometieron a cirugía después del tratamiento, en comparación con el 69 por ciento en el grupo de control. «Es posible que más personas se operen con este enfoque porque este tratamiento es más efectivo para reducir el tamaño del tumor, sin agregar mucha toxicidad»,
La quimioterapia consiste en tratar el cáncer con fármacos que son más tóxicos para las células cancerosas que para las sanas. Los fármacos empleados tradicionalmente envenenan las células que se están dividiendo, interrumpiendo así la duplicación del ADN o la separación de cromosomas recién formados. Como en el caso de la radioterapia, las células normales tienen mayor capacidad de recuperarse del daño que las cancerosas. Por esta razón la quimioterapia a menudo se administra en las dosis máximas tolerables, con los consiguientes efectos secundarios relativos a la regeneración celular, tales como la pérdida de mucosa oral y gastrointestinal, cabello, piel y uñas.
Una meta importante de la investigación actual es desarrollar fármacos dirigidos a las células cancerosas pero basados en la dependencia de dichas células de las mutaciones oncogénicas que contienen (Sawyers 2004) (fig. 1). Estas terapias dirigidas no son diferentes de muchos fármacos empleados para otro tipo de enfermedades. Los fármacos dirigidos contra el cáncer buscan alcanzar mayor efectividad terapéutica con menos efectos secundarios. Su utilización permite que los niveles de radioterapia aplicados sean menores y menores también por tanto sus efectos secundarios. Las terapias dirigidas incluyen compuestos químicos que por lo general actúan inhibiendo la actividad enzimática de los productos de los oncogenes, y anticuerpos monoclonales que actúan bloqueando el receptor oncógeno de la superficie de una célula o matando la célula cancerosa, destruyendo sus anticuerpos.
Las terapias dirigidas surgieron en la década de 1990 como consecuencia directa de la identificación de genes cancerosos críticos. La capacidad para analizar tumores molecularmente está revolucionando la clasificación, la prognosis y el tratamiento del cáncer. Las investigaciones en este campo continúan siendo intensas y prometedoras. Entre los anticuerpos monoclonales, el anticuerpo anti-HER2 trastuzumab (Herceptin TM) resulta efectivo contra un subtipo de carcinomas de mama que contienen un oncogén HER2 activado (Hudis 2007; Shawver et al. 2002). El anticuerpo anti-CD20 rituxiab (TiruxanTM) se usa para tratar linfomas de células B que presentan el antígeno CD20 (Cheson y Leonard 2008), y el anticuerpo anti-EGFR cetuximab (ErbituxTM) se usa en cáncer de colon avanzado (Mendelsohn y Baselga 2006) (fig. 4).
Entre los compuestos químicos dirigidos, el imatinib (GleevecTM), un inhibidor de pequeñas moléculas del oncogén BCR-ABL quinasa se emplea con éxito contra leucemias causadas por este oncogén (Schiffer 2007). El inhibidor EGFR erlotinib (TarcevaTM) se usa en carcinomas de pulmón causados por un EGFR mutante (Ciardelloy Tortora 2008). Además, aunque diferentes subtipos de cáncer en un órgano determinado pueden experimentar clases de mutaciones diferentes, ciertos subtipos de cáncer en órganos distintos pueden, sorprendentemente, compartir mutaciones. Como resultado de ello un mismo fármaco puede ser efectivo en tumores de tipo molecular localizados en órganos distintos. Una clase relacionada de compuestos anticáncer son los inhibidores de la angiogénesis, que previenen la formación de capilares sanguíneos que alimentan los tumores. Algunos, tales como el anticuerpo monoclonal bevacizumab (AvastinTM) son ya de uso clínico (Ferrara 2002) (fig. 4), aunque con escaso éxito, debido a que las células cancerosas tienen múltiples maneras de estimular la angiogénesis (Meyerhardt y Mayer 2005). Las investigaciones actuales en este campo por tanto se centran en identificar combinaciones de inhibidores de la angiogénesis que sean efectivas (Bergers y Hanahan 2008).
Siguen siendo muchas las barreras para mejorar el tratamiento del cáncer, lo que conforma la necesidad de emprender nuevas direcciones. ¿Qué cambios podemos prever en el campo de la oncología? En un futuro no muy lejano, el perfil tumoral de un paciente podría incluir no sólo gradación histopatológica e información sobre el estado de las mutaciones oncogénicas comunes, sino también un completo retrato molecular del tumor (Massagué 2007; Nevins y Pottu 2007). Avances recientes en el diseño de perfiles moleculares de tumores han llevado al descubrimiento de perfiles de expresión de genes que permiten clasificar mejor los tumores en subtipos diferenciados, predecir los riesgos y la localización de metástasis con mayor precisión e identificar mejor los objetivos terapéuticos (Bild et al. 2006; Fan et al. 2006; Minn et al. 2005; Padua et al. 2008; Van’t Veer et al. 2002, Van de Vijver et al. 2002). Un perfil de «prognosis desfavorable» de setenta genes (MammaPrint) y un conjunto de veintiún genes no coincidentes de «recurrencia» (Oncotype Dx) han sido transformados en fármacos comerciales que ayudan a los médicos a tomar decisiones, evitando mastectomías en pacientes de cáncer de mama como tumores que presentan una prognosis favorable después de la quimioterapia. Los genes de dichos perfiles pueden ser entonces examinados directamente para medir su capacidad de formar metástasis y tratados con terapia dirigida para reducir la actividad metastásica de las células cancerosas (Gupta et al. 2007). Los tratamientos farmacológicos para pacientes de cáncer pueden incluir combinaciones individualizadas dirigidas a subtipos específicos de tumor y a sus metástasis. La mejora de los biomarcadores de respuesta a los fármacos en pacientes ayudará a evaluar con mayor precisión la reacción de éstos a terapias dirigidas (Sawyers 2008).
Los nuevos conocimientos acerca de las bases moleculares, genéticas y celulares del desarrollo y la progresión del cáncer traen consigo nuevas oportunidades de mejorar y expandir nuestra capacidad de prevenir, detectar y tratar esta enfermedad. Trabajando en estrecha colaboración, científicos y médicos pueden generar y aplicar los conocimientos necesarios para hacer del cáncer una enfermedad crónica más en las próximas décadas.
BIBLIOGRAFÍA
American Cancer Society. Cancer facts and fi-gures 2008. http://www.cancer.org/, 2008.
Bergers, G., y D. Hanahan. «Modes of resistance to anti-angiogenic therapy», Nature Reviews Cancer 8 (2008): 592-603.
Bierie, B., y H. L. Mose. «Tumor microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer», Nature Reviews Cancer 6 (2008): 506-520. (*)
Bild, A. H., G. Yao, J. T. Chang, Q. Wang, A. Potti, D. Chasse, M. B. Joshi, D. Harpole, J. M. Lancaster, A. Berchuck et al. «Oncogenic pathway signatures in human cancers as a guide to targe-ted therapies». Nature 439 (2006): 353-357.
Billingsley, K.G., M. E. Burt, E. Jara, R. J. Ginsberg, J. M. Woodruff, D. H. Leung y M. F. Brennan. «Pulmonary metastases from soft tissue sarcoma: analysis of patterns of diseases and postmetastasis survival». Annals of Surgery 229, (1999): 602-610; Discussion (1999): 610-602.
Blasco, M. A. «The epigenetic regulation of mammalian telomeres». Nature Reviews Genetics 8 (2007): 299-309.
Boshoff, C. y R. Weiss. «AIDS-related malignancies». Nature Reviews Cancer 2 (2002): 373-382.
Cano, A., M. A. Pérez-Moreno, I. Rodrigo, A. Locascio, M. J. Blanco, M. G. del Barrio, F. Portillo y M. A. Nieto. «The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression». Nature Cell Biology 2 (2000): 76-83.
Cheson, B. D. y J. P. Leonard. «Monoclonal antibody therapy for B-cell non-Hodgkin’s lymphoma». The New England Journal of Medicine 359 (2008): 613-626.
Cheung, T. K., H. H. Xia y B. C. Wong. «Helicobacter pylori eradication for gastric cancer prevention». Journal of Gastroenterology 42, supl 17 (2007): 10-15.
Christofori, G. «New signals from the invasive front». Nature 441 (2006): 444-450.
Ciardiello, F. y G. Tortora, G. «EGFR antagonists in cancer treatment». The New England Journal of Medicine 358 (2008): 1.160-1.174.
Clark, E. A., T. R. Golub, E. S. Lander y R. O. Hynes. «Genomic analysis of metastasis reveals an essential role for RhoC». Nature 406 (2002): 532-535.
Clarke, M. F. y M. Fuller. «Stem cells and cancer: two faces of eve». Cell 124, (2006): 1.111-1.115.
Classon, M. y E. Harlow. «The retinoblastoma tumour suppressor in development and cancer». Nature Reviews Cancer 2 (2002): 910-917.
Danaei, G., S. Vander Hoorn, A. D. López, C. J. Murray y M. Ezzati. «Causes of cancer in the world: comparative risk assessment of nine behavioural and environmental risk factors». Lancet 366 (2005): 1.784-1.793.
De la Chapelle, A. «Genetic predisposition to colorectal cancer». Nature Reviews Cancer 4 (2004): 769-780.
Esteller, M. «Cancer epigenomics: DNA methy-lomes and histone-modification maps». Nature Reviews Genetics 8 (2007): 286-298.
Fan, C., D. S. Oh, L. Wessels, B. Weigelt, D. S. Nuyten, A. B. Nobel, L. J. Van’t Veer y C. M. Perou. «Concordance among gene-expression-based predictors for breast cancer». The New England Journal of Medicine 355 (2006): 560-569.
Ferrara, N. «VEGF and the quest for tumour angiogenesis factors». Nature Reviews Cancer 2 (2002): 795-803.
Fidler, I. J. «The pathogenesis of cancer metastasis: the «seed and soil» hypothesis revisited». Nature Reviews Cancer 3 (2003): 453-458.
Fodde, R., R. Smits y H. Clevers. «APC, signal transduction and genetic instability in colorectal cancer». Nature Reviews Cancer 1 (2001): 55-67.
Gavrilovic, I. T. y J. B. Posner. «Brain metastases: epidemiology and pathophysiology». Neurooncology 75 (2005): 5-14.
Gupta, G. P. y J. Massagué. «Cancer metastasis: building a framework». Cell 127 (2006): 679-695.
—, D. X. Nguyen, A. C. Chiang, P. D. Bos, J. Y. Kim, C. Nadal, R. R. Gomis, K. Manova-Todorova y J. Massagué. «Mediators of vascular remo-delling co-opted for sequential steps in lung metastasis». Nature 446 (2007): 765-770.
Halazonetis, T. D., V. G. Gorgoulis y J. Bartek. «An oncogene-induced DNA damage model for cancer development». Science 319 (2008): 1.352-1.355.
Hanahan, D. y R. A Weinberg. «The hallmarks of cancer». Cell 100 (2000): 57-70.
Hess, K. R., G. R. Varadhachary, S. H. Taylor, W. Wei, M. N. Raber, R. Lenzi y J. L. Abbruzzese. «Metastatic patterns in adenocarcinoma». Cancer 106 (2006): 1.624-1.633.
Hudis, C. A. «Trastuzumab-mechanism of action and use in clinical practice». The New England Journal of Medicine 357 (2007): 39-51.
Husemann, Y., J. B. Geigl, F. Schubert, P. Musiani, M. Meyer, E. Burghart, G. Forni, R. Eils, T. Fehm, G. Riethmuller et al. «Systemic spread is an early step in breast cancer». Cancer Cell 13 (2008): 58-68.
Jemal, A., T. Murray, E. Ward, A. Samuels, R. C. Tiwari, A. Ghafoor, E. J. Feuer y M. J. Thun. «Cancer statistics, 2005». CA: a cancer journal for clinicians 55 (2005): 10-30.
Joyce, J. A. «Therapeutic targeting of the tumor microenvironment». Cancer Cell 7 (2005): 513-520.
Kaelin, W. G. «The von Hippel-Lindau tumor suppressor protein: roles in cancer and oxygen sensing». Cold Spring Harbor Symposia in Quantitative Biology 70 (2005): 159-166.
Kang, Y., P. M Siegel, W. Shu, M. Drobnjak, S. M. Kakonen, C. Cordon-Cardo, T. A. Guise y J. Massagué. «A multigenic program media-ting breast cancer metastasis to bone». Cancer Cell 3 (2003): 537-549.
Karin, M. «Nuclear factor-kappaB in cancer development and progression». Nature 441 (2006): 431-436.
Karpozilos, A. y N. Pavlidis. «The treatment of cancer in Greek antiquity». European Journal of Cancer 40 (2004): 2.033-2.040.
Kim, M., J. D. Gans, C. Nogueira, A. Wang, J. H. Paik, B. Feng, C. Brennan, W. C. Hahn, C. Cordon-Cardo, S. N. Wagner et al. «Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene». Cell 125 (2006): 1.269-1.281.
Klein, C. A., T. J. Blankenstein, O. Schmidt-Kittler, M. Petronio, B. Polzer, N. H. Stoecklein y G. Riethmuller. «Genetic heterogeneity of single disseminated tumour cells in minimal residual cancer». Lancet 360 (2002): 683-689.
Leiter, U., F. Meier, B. Schittek y C. Garbe. «The natural course of cutaneous melanoma». Journal of Surgical Oncology 86 (2004): 172-178.
Lewis, C. E. y J. W. Pollard. «Distinct role of macrophages in different tumor microenvironments». Cancer Research 66 (2006): 605-612.
Malumbres, M. y M. Barbacid. «Cell cycle kinases in cancer». Current Opinion in Genetics and Development 17 (2007): 60-65.
Martin, R. W., P. P. Connell y D. K. Bishop. «The Yin and Yang of treating BRCA-deficient tumors». Cell 132 (2008): 919-920.
Massagué, J. «G1 cell-cycle control and cancer». Nature 432 (2004): 298-306.
—, «Sorting out breast-cancer gene signatures». The New England Journal of Medicine 356 (2007): 294-297.
—, «TGFbeta in Cancer». Cell 134 (2008): 215-230.
Melo, J. V. y D. J. Barnes. «Chronic myeloid leukaemia as a model of disease evolution in human cancer». Nature Reviews Cancer 7 (2007): 441-453.
Mendelsohn, J. y J. Baselga. «Epidermal growth factor receptor targeting in cancer». Seminars in Oncology 33 (2006): 369-385.
Meyerhardt, J. A. y R. J. Mayer. «Systemic therapy for colorectal cancer». The New England Journal of Medicine 352 (2005): 476-487.
Minn, A.J., Gupta, G.P., Padua, D., Bos, P., Nguy-en, D.X., Nuyten, D., Kreike, B., Zhang, Y., Wang, Y., Ishwaran, H., et al. (2007). Lung metastasis genes couple breast tumor size and metastatic spread. Proceedings of the National Academy of Sciences USA 104, 6740-6745.
Minn, A. J., G. P. Gupta, P. M. Siegel, P. D. Bos, W. Shu, D. D. Giri, A. Viale, A. B. Olshen, W. L. Gerald y J. Massagué. «Genes that mediate breast cancer metastasis to lung». Nature 436 (2005): 518-524.
Mueller, M. M. y N. E. Fusenig. «Friends or foes – bipolar effects of the tumour stroma in cancer». Nature Reviews Cancer 4 (2004): 839-849.
Nevins, J. R. y A. Potti. «Mining gene expression profiles: expression signatures as cancer phenotypes». Nature Reviews Genetics 8 (2007): 601-609.
Norton, L. J. Massagué. «Is cancer a disease of self-seeding?». Nature Medicine 12 (2006): 875-878.
Nowell, P. C. «The clonal evolution of tumor cell populations». Science 194 (1976): 23-28.
Olsson, A. K., A. Dimberg, J. Kreuger y L. Claesson-Welsh. «VEGF receptor signalling – in control of vascular function». Nature Revies Molecular Cell Biology 7 (2006): 359-371.
Padua, D., X. H. Zhang, Q. Wang, C. Nadal, W. L. Gerald, R. R. Gomis y J. Massagué. «TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4». Cell 133 (2008): 66-77.
Paget, S. «The distribution of secondary growths in cancer of the breast». Lancet 1 (1889): 571-573.
Parato, K. A., D. Senger, P. A. Forsyth y J. C. Bell. «Recent progress in the battle between oncolytic viruses and tumours». Nature Reviews Cancer 5 (2005): 965-976.
Perl, A. K., P. Wilgenbus, U. Dahl, H. Semb y G. Christofori. «A causal role for E-cadherin in the transition from adenoma to carcinoma». Nature 392 (1998): 190-193.
Pouyssegur, J., F. Dayan y N. M. Mazure. «Hypoxia signalling in cancer and approaches to enforce tumour regression». Nature 441 (2006): 437-443.
Roden, R., A. Monie y T. C. Wu. «The impact of preventive HPV vaccination». Discovery Medicine 6 (2006): 175-181.
Rustgi, A. K. «The genetics of hereditary colon cancer». Genes and Development 21 (2007): 2.525-2.538.
Sawyers, C. L. «Targeted cancer therapy». Nature 432 (2004): 294-297.
—, «The cancer biomarker problem». Nature 452 (2008): 548-552.
Schiffer, C. A. «BCR-ABL tyrosine kinase inhibitors for chronic myelogenous leukemia». The New England Journal of Medicine 357 (2007): 258-265.
Schmidt-Kittler, O., T. Ragg, A. Daskalakis, M. Granzow, A. Ahr, T. J. Blankenstein, M. Kaufmann, J. Diebold, H. Arnholdt, P. Muller et al. «From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression». Proceedings of the National Academy of Sciences U S A 100 (2003): 7.737-7.742.
Shawver, L. K., D. Slamon y A. Ullrich. «Smart drugs: tyrosine kinase inhibitors in cancer therapy». Cancer Cell 1 (2002): 117-123.
Sweet-Cordero, A., S. Mukherjee, A. Subramanian, H. You, J. J. Roix, C. Ladd-Acosta, J. Mesirov, T. R. Golub y T. Jacks. «An oncogenic KRAS2 expression signature identified by cross-species gene-expression analysis». Nature Genetics 37 (2005): 48-55.
Thiery, J. P. «Epithelial-mesenchymal transitions in tumour progression». Nature Reviews Cancer 2 (2002): 442-454.
Van‘t Veer, L. J., H. Dai, M. J. Van de Vijver, Y. D. He, A. A. Hart, M. Mao, H. L. Peterse, K. Van der Kooy, M. J. Marton, A. T. Witteveen et al. «Gene expression profiling predicts clinical outcome of breast cancer». Nature 415 (2002): 530-536.
Van de Vijver, M. J., Y. D. He, L. J. Van’t Veer, H. Dai, A. A. Hart, D. W. Voskuil, G. J. Schreiber, J. L. Peterse, C. Roberts, M. J. Marton et al. «A gene-expression signature as a predictor of survival in breast cancer». The New England Journal of Medicine 347 (2002): 1.999-2.009.
Vogelstein, B., E. R. Fearon, S. R. Hamilton, S. E. Kern, A. C. Preisinger, M. Leppert, Y. Nakamura, R. White, A. M. Smits y J. L. Bos. «Genetic alterations during colorectal-tumor development». The New England Journal of Medicine 319 (1988): 525-532.
—, y K. W. Kinzler. «Cancer genes and the pathways they control». Nature Medicine 10 (2004): 789-799.
Vousden, K. H. y D. P. Lane. «p53 in health and disease». Nature Reviews Molecular Cell Biology 8 (2007): 275-283.
Walsh, T. y M. C. King. «Ten genes for inherited breast cancer». Cancer Cell 11 (2007): 103-105.
Wang, C., Y. Yuan y R. H. Hunt. «The association between Helicobacter pylori infection and early gastric cancer: a meta-analysis». The American Journal of Gastroenterology 102 (2007): 1.789-1.798.
Wang, J., R. Loberg y R. S. Taichman. «The pivotal role of CXCL12 (SDF-1)/CXCR4 axis in bone metastasis». Cancer Metastasis Rev 25 (2006): 573-587.
Wang, W. «Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins». Nature Reviews Genetics 8 (2007): 735-748.
Weinberg, R. A. The biology of cancer. New York: Garland Science. 2007.
Welcsh, P. L. y M. C. King. «BRCA1 and BRCA2 and the genetics of breast and ovarian cancer». Human Molecular Genetics 10 (2001): 705-713.
Woodman, C. B., S. I. Collins y L. S. Young. «The natural history of cervical HPV infection: unresolved issues». Nature Reviews Cancer 7 (2007): 11-22.
WorldHeatlhOrganization. Cancer. Fact Sheet No 297. 2008. http://www.who.int/mediacentre/factsheets/fs297/en/index.html.
Yang, J., S. A. Mani, J. L. Donaher, S. Ramaswamy, R. A. Itzykson, C. Come, P. Savagner, I. Gitelman, A. Richardson y R. A. Weinberg. «Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis». Cell 117 (2004): 927-939.
Young, L. S. y A. B. Rickinson. «Epstein-Barr virus: 40 years on». Nature Reviews Cancer 4 (2004): 757-768.
Zur Hausen, H. «Viruses in human cancers». European Journal of Cancer 35 (1999): 1174-1181.


 Lo que movía a quienes padecían el síndrome de Hubris era una ambición sin límites, con un comportamiento temerario e insolente que les llevaba a pensar que podrían conseguir mucho más allá de lo que el destino les había previsto.
Lo que movía a quienes padecían el síndrome de Hubris era una ambición sin límites, con un comportamiento temerario e insolente que les llevaba a pensar que podrían conseguir mucho más allá de lo que el destino les había previsto.








 Se cree que el Homo erectus fue el primer homínido en conocer el fuego.
Se cree que el Homo erectus fue el primer homínido en conocer el fuego.
 El Homo erectus es un buen candidato a ser la especie previa al Homo sapiens.
El Homo erectus es un buen candidato a ser la especie previa al Homo sapiens. Muchos Homo erectus pueden haber convivido con los primeros Homo sapiens.
Muchos Homo erectus pueden haber convivido con los primeros Homo sapiens.





 Elena Garralda, jefa del Grupo de Desarrollo Clínico Precoz de Fármacos del Vall d’Hebron Instituto de Oncología (VHIO). Foto: VHIO.
Elena Garralda, jefa del Grupo de Desarrollo Clínico Precoz de Fármacos del Vall d’Hebron Instituto de Oncología (VHIO). Foto: VHIO.

 DURMIENDO LA TAJA
DURMIENDO LA TAJA
 Expertos señalan la importancia de cortar las cadenas de transmisión de la viruela del mono. Foto:
Expertos señalan la importancia de cortar las cadenas de transmisión de la viruela del mono. Foto: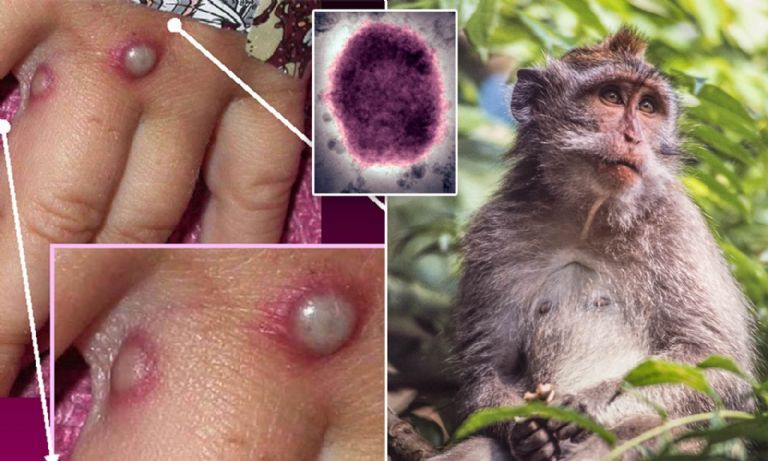 Esta semana, con la detección el 14 de mayo de dos casos en Reino Unido y 30 casos confirmados ya en España, el mayor número fuera de África, han sonado todas las alarmas y la Organización Mundial de la Salud (OMS) prevé una reunión extraordinaria para la próxima semana. ¿Hay motivos serios de preocupación?
Esta semana, con la detección el 14 de mayo de dos casos en Reino Unido y 30 casos confirmados ya en España, el mayor número fuera de África, han sonado todas las alarmas y la Organización Mundial de la Salud (OMS) prevé una reunión extraordinaria para la próxima semana. ¿Hay motivos serios de preocupación? Daniel López Acuña, epidemiólogo y ex director de Acción Sanitaria de la OMS.
Daniel López Acuña, epidemiólogo y ex director de Acción Sanitaria de la OMS.