TALAMO
El término tálamo deriva de la palabra griega “thalamos” que significa “cámara interna” o“lecho nupcial”. Galeno (130-200 d. C.)
El sistema nervioso central (SNC) esta integrado por dos grandes regiones, el encéfalo y la médula espinal.
El encéfalo, constituido por todas las estructuras del SNC situadas dentro del cráneo, está formado por el cerebro (hemisferios cerebrales y diencéfalo) y el tronco encefálico (mesencéfalo, protuberancia /cerebelo y bulbo raquídeo).
El diencéfalo está constituido por un conjunto de estructuras nerviosas situadas alrededor del tercer ventrículo y recubiertas en gran parte por los hemisferios cerebrales. Macroscópicamente puede verse en la región basal del encéfalo donde podemos localizar las estructuras diencefálicas inferiores, las que forman el suelo del diencéfalo: quiasma óptico, hipófisis y cuerpo mamilar. En contacto directo con la base del cráneo, sólo separado del hueso por las meninges y espacios entre membranas. En la parte más caudal el diencéfalo se continúa con estructuras mesencefálicas y a derecha e izquierda con los hemisferios cerebrales.
El tálamo
La estructura mayor del diencéfalo compuesta por dos ovoides unidos por una comisura. Y posiblemente la mas importante y vital.
Todo pasa por el Talamo, menos el Olfatorio, que se le acerca pero no lo cruza.
El talamo califica y corrige todos los estimulos que percibimos y los envia a la corteza cerebral y a nucleos del diencéfalo.
El talamo lo sabe todo
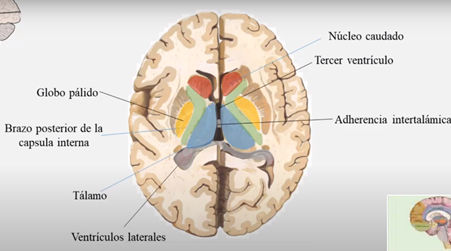
Las caras mediales de ambos tálamos están unidas entre sí por un conjunto de fibras intertalámicas conocidas con el nombre de masa intermedia, adherencia intertalámica o comisura gris intertalámica. La cara lateral de forma cóncava, está separada del núcleo lenticular por el brazo posterior de la cápsula interna. 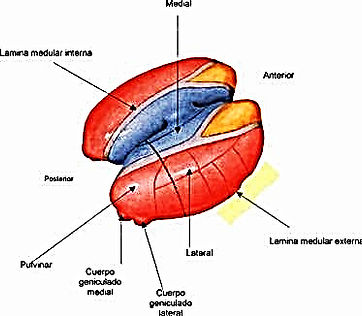
Neuronas del Tálamo.
En el tálamo existen dos tipos de neuronas desde un punto de vista funcional:
1.- neuronas principales o de proyección (transmiten información fuera del tálamo), las cuales representan cerca del 75% de la población neuronal total; y
2.- interneuronas locales o de circuitos locales (pueden recibir información de las mismas fuentes que las neuronas principales pero sólo entran en contacto con células talámicas que participan en la misma etapa de procesamiento), las cuales constituyen alrededor del 25%. 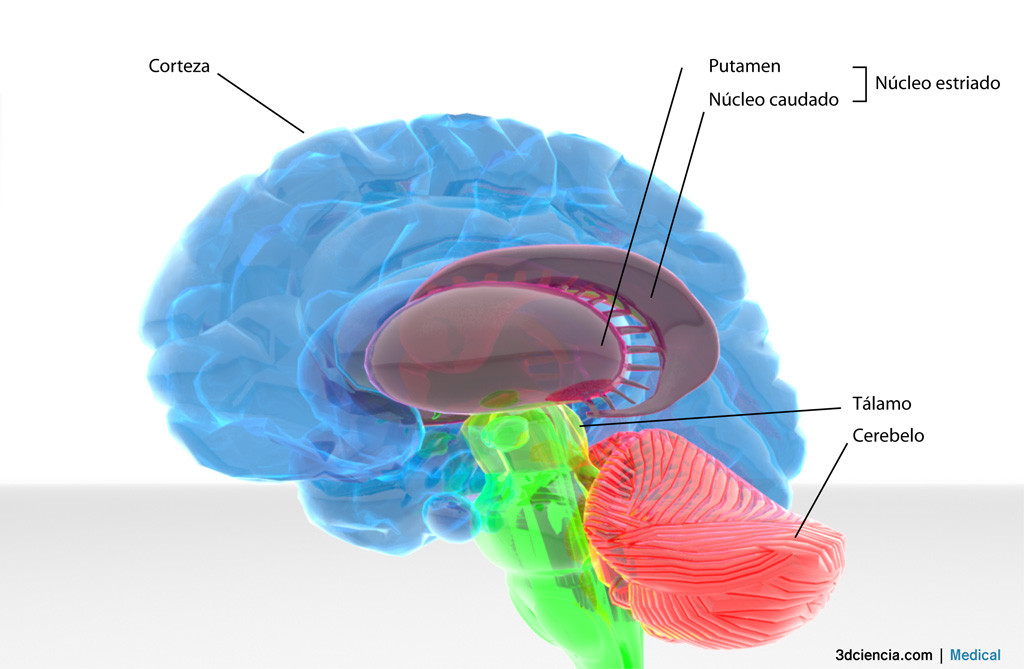
Las neuronas principales envían sus axones a la corteza cerebral, donde liberan un neurotransmisor excitatorio (glutamato generalmente) para activar las neuronas corticales. El glutamato y el aspartato son neurotransmisores excitatorios presentes en las terminaciones corticotalámicas y cerebelosas y en las neuronas de proyección talamocortical. Una excepción lo constituyen las aferencias subcorticales de los núcleos grises de la base que son GABA-érgicas, inhibitorias.
Las neuronas de circuitos locales liberan ácido gammaaminobutírico (GABA) en las células de proyección para inhibirlas. Este neurotransmisor inhibitorio se localiza en las terminaciones que provienen del globo pálido, en las neuronas de circuitos locales y en las de proyección del núcleo reticular y cuerpo geniculado lateral. Son proyecciones GABA-érgicas las principales proyecciones del segmento palidal medial hacia el ventral anterior (parvocelular) y el ventral lateral (pars oralis) y las proyecciones de la parte reticular de la sustancia negra al núcleo ventral anterior (magnocelular) y dorsomedial (paralaminar). Estas aferencias desempeñan un papel fundamental en la función motora [2]. Las neuronas GABA-érgicas han sido identificadas en todas las láminas del cuerpo geniculado lateral, siendo más abundantes en las láminas 1 y 2 (magnocelulares).
Las aferencias procedentes de regiones subcorticales y de la corteza cerebral que se dirigen hasta los núcleos talámicos, excitan (despolarizan) a las neuronas de proyección e interneuronas locales de dichos núcleos. A su vez las neuronas de circuitos locales inhiben (hiperpolarizan) a las neuronas de proyección, el neurotransmisor utilizado es el GABA. Así las aferencias hacia el tálamo influyen sobre las neuronas de proyección (tálamocorticales) a través de dos vías: una excitatoria directa y una inhibitoria indirecta (por medio de las neuronas de circuitos locales). Las neuronas de circuitos locales modulan la actividad de las neuronas de proyección, las cuales envían sus axones a los destinos extratalámicos. Además las células de proyección envían una rama colateral a las neuronas del núcleo reticular talámico, el cual contiene el neurotransmisor inhibitorio GABA, actuando como neuronas de circuitos locales. Las células del núcleo reticular talámico envían ramas axónicas a las neuronas de proyección y de circuitos locales por lo que ambas son inhibidas. La corteza cerebral, la cual recibió proyecciones aferentes excitatorias de las células talámicas de proyección, envía axones excitatorios de regreso a todos los tipos celulares talámicos, por lo que las aferencias corticales activan a las neuronas de proyección así como a las inhibitorias de circuitos locales y del núcleo reticular.
De esta forma el tálamo no sólo es un simple relevo de información entre los centros aferentes y la corteza, sino que es el encargado del procesamiento de la información, influyendo por tanto sobre las funciones corticales [3].
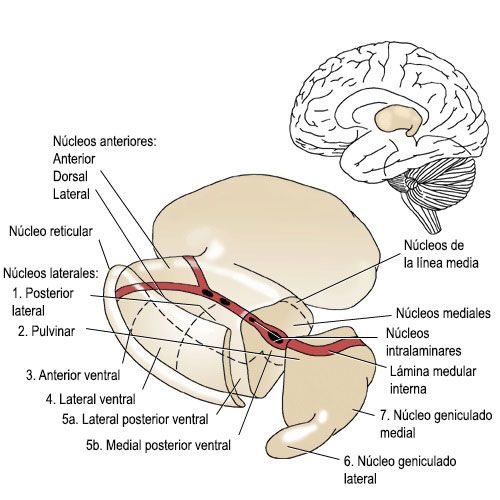
Grupos nucleares talámicos
En el tálamo,se han identificado hasta 50 núcleos talámicos [4], varios de los cuales son subdivisiones microscópicas. La nomenclatura de los núcleos talámicos es muy compleja y en algunos casos se desconocen sus conexiones y la significación funcional de los más pequeños [2].
Su complejidad ha permitido diferentes formas de clasificar sus núcleos y las funciones y proyecciones de estos. 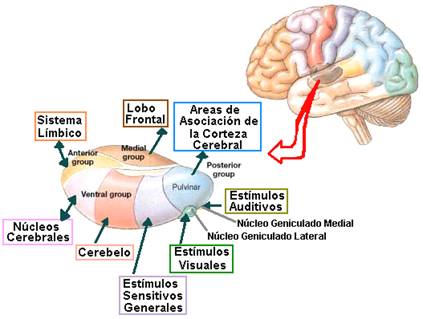
I- CLASIFICACIÓN DESDE UNA PERSPECTIVA EVOLUTIVA.
II- CLASIFICACIÓN ANATOMO-FUNCIONAL.
III- CLASIFICACIÓN BASADA EN CRITERIOS CITOARQUITECTÓNICOS.
IV- CLASIFICACIÓN TENIENDO EN CUENTA LAS CONEXIONES.
V- CLASIFICACIÓN BASADA EN LAS CARACTERÍSTICAS COMPARTIDAS DE CONECTIVIDAD DE FIBRAS Y FUNCIONES.
Desde una perspectiva evolutiva, los Núcleos de la línea media tienen escaso desarrollo en seres humanos y son difíciles de delimitar, localizados en la sustancia gris periventricular, por encima del surco hipotalámico. Mantiene estrechas relaciones con el hipotálamo, los núcleos intralaminares y el núcleo dorsomedial .
Entre los núcleos de la línea a destacar el:
– Núcleo paratenial
– Núcleo paraventricular
– Núcleo reuniens
– Núcleo romboide
Participan en las emociones, la memoria y la funciones autonómica.
Núcleos intralaminares
Constituyen una numerosa serie de acúmulos neuronales situados en el espesor de la lámina medular interna del tálamo (figura 3). Los dos núcleos principales desde el punto de vista funcional en humanos son el:
– Núcleo centromediano
– Núcleo parafascicular
Otros núcleos que se localizan más hacia la región rostral incluye el:
– Núcleo paracentral
– Núcleo central lateral
– Núcleo central medial.
Los núcleos intralaminares, elementos que funcionan, llevando y modulando sensibilidades plurales inespesificas muy primitivas
Los núcleos intralaminares influyen sobre la actividad cortical a través de otros núcleos talámicos. Desempeñan una función global activadora, debida a sus conexiones múltiples extratalámicas y corticales y una función específica. Las conexiones que estos núcleos mantienen con el putamen y el caudado contribuyen al control motor subcortical.
Núcleos reticulares 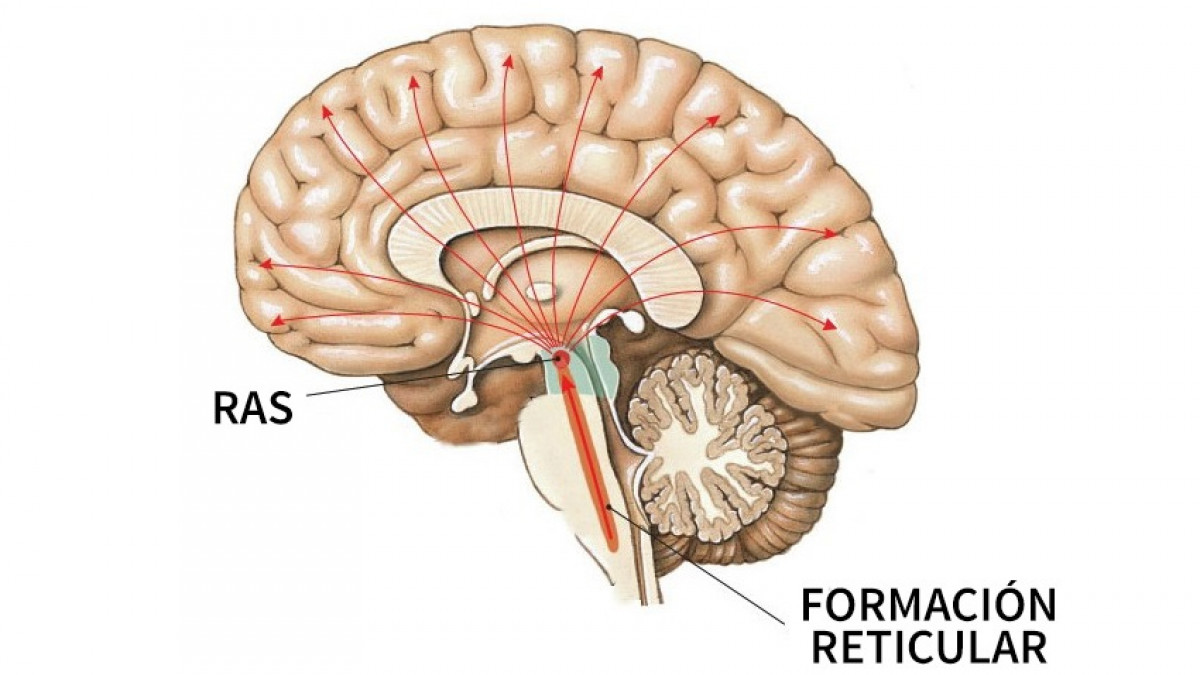
Recubren el polo anterior y la cara lateral del tálamo, del que queda separado por una delgada hoja de sustancia blanca, la lámina medular externa. Se localizan entre la lámina medular externa y la cápsula interna
Entre estos núcleos reticulares destacamos:
– núcleo reticular. es una delgada lámina de sustancia gris vertical, que se apoya sobre la cara externa del tálamo.
– núcleos reticulares de la línea media. pequeñas masas grises adosadas a la cara medial del tálamo.
– núcleo centro mediano. es un núcleo grande situado en el espesor de la lámina medular externa.
los núcleos intralaminares, reticular y de la línea media considerados habitualmente como inespecíficos, están relacionados con el despertar, control motor y la conciencia de las experiencias sensoriales.
CLASIFICACIÓN TENIENDO EN CUENTA LAS CONEXIONES
Se diferencian dos grandes grupos nucleares talámicos: específicos e inespecíficos.
1.- Núcleos específicos: aquellos que tienen una relación específica y selectiva con una parte concreta de la neocorteza. Es decir, son eslabones intermedios en el procesamiento de la información hasta la corteza y funcionan de un modo complejo, colaborando en la integración, selección, procesamiento y transmisión hacia el cortéx cerebral. Cada uno de estos núcleos recibe proyecciones desde el área de la corteza cerebral a la que ha enviado sus eferencias, es decir existe una reciprocidad entre estos núcleos y la corteza cerebral. Se conocen también como núcleos corticodependientes.
Dentro de estos núcleos específicos se diferencian dos tipos:
– Núcleos específicos de relevo: reciben aferencias directas de las áreas subcorticales y se proyectan a la capa IV de la corteza cerebral. Este grupo lo integran los siguientes núcleos: grupo nuclear anterior, núcleo VA, núcleo VL, VP, CGL y CGM. Según Martín [10] los núcleos de relevo son esenciales para todas las funciones cerebrales, y cada uno de ellos desempeña un papel diferente en la percepción, la volición o la cognición, transmitiendo información desde estructuras subcorticales concretas a una porción limitada del córtex.
– Núcleos específicos de asociación: reciben aferencias corticales y de otros núcleos talámicos y a su vez ellos se proyectan a diversas capas corticales (I, III y VI). Este grupo lo integran los siguientes núcleos: DL, LP, DM y pulvinar.
2.- Núcleos inespecíficos: se proyectan a varias regiones corticales y subcorticales. Son los núcleos de la línea media, intralaminares y reticulares. Para estos núcleos la corteza cerebral no es su lugar de proyección principal. Se ha comprobado que los dos primeros envían proyecciones a regiones subcorticales precisas, por lo que no son tan inespecíficos como se pensaba [2]. Actúan en el despertar y regulando la excitabilidad de las regiones más amplias de la corteza cerebral [10]. Se le denomina también núcleos corticoindependientes.
V- Clasificación basada en las características compartidas de conectividad de fibras y funciones
Según Afifi y Bergman [6] en general se utilizan dos sistema de nomenclatura:
A).- Núcleos talámicos agrupados en tres categorías generales teniendo en cuenta la conectividad de las fibras.
1.- De modalidad específica: comparten las siguientes características:
a.- reciben aferencias directas de tractos ascendentes largos relacionados con información somatosensorial, visual y auditiva (VPL y VPM, CGL y CGM) o cualquier proceso de información derivado de los núcleos grises de la base (VA, VL), el cerebelo (VL) o el sistema límbico (núcleo anterior y DL).
b.- tienen conexiones recíprocas con áreas corticales bien definidas (área somatosensorial primaria, auditiva y visual, áreas premotoras y motoras primaria y giro cingulado)
c.- Se degeneran mediante la ablación de las áreas corticales específicas a las que se proyectan.
2.- Multimodal asociativo: no reciben aferencias directas de los tractos ascedentes largos y se proyectan áreas de asociación en los lóbulos frontales, parietal y temporal. Incluyen, el DM y el complejo nuclear pulvinar-lateral posterior.
Tienen la siguientes características en común: 1- No reciben aferencias de los tractos ascendentes largos; 2- la mayor parte de sus aferencias provienen de otros núcleos talámicos; 3- su proyección principal se dirige a las áreas de asociación de la corteza cerebral.
3.- Inespecíficos y reticular: se caracterizan por proyecciones corticales indirectas difusas y amplias y por aferencias de la formación reticular troncoencefálica. Incluyen los núcleos intralaminares, de la línea media y reticulares.
B).- Teniendo en cuenta la función que desempeñan
1.- Motores: reciben aferencia motoras de los núcleos grises de la base (VA) o el cerebelo (VL) y se proyecta a las cortezas premotoras y motoras primarias.
2.- Sensitivos: recibe aferencias de los sistemas ascendentes somatosensoriales (VPL y VPM), auditivo (CGM) y visual (CGL).
3.- Límbicos: se relacionan con estructuras límbicas (cuerpos mamilares, hipocampo, giro del cíngulo).
4.- Asociativos: se corresponden con los núcleos multimodales asociativos.
5.- Inespecífico y reticular: corresponden a la misma categoría del otro sistema de nomenclatura.
Conexiones tálamo-corticales y cortico-talámicas
La organización en el seno de la corteza cerebral de las proyecciones tálamo-corticales y cortico-talámicas y las propiedades neurofisiológicas de las fibras que ascienden o descienden a o desde la corteza cerebral son la base de las complejas relaciones entre los diferentes núcleos talámicos y la corteza cerebral [11].
Fue Lorente de Nó [12] quien describió las aferencias tálamo-corticales como: fibras tálamo-corticales específicas y fibras tálamo-corticales inespecíficas. Las primeras tienen su origen en los núcleos específicos del tálamo, sinaptan en la capa IV de la corteza y son portadoras de información de la sensibilidad general y especial (excepto la olfativa). Las segundas son fibras ascendentes con colaterales fundamentalmente a las capas I, II y VI. Estas vías inespecíficas están relacionadas con las vías tálamo-corticales difusas, procedentes de los núcleos de la línea media e intralaminares hacia el córtex cerebral [13,14] y relacionados con los mecanismos de arousal.
Macchi [13] en estudios realizados en gatos señala que existen cuatro tipos de conexiones tálamo-corticales, en función de la difusión de sus eferencias.
La primera incluye todos los núcleos que envían sus proyecciones sobre un área anatomofuncional homogénea de la corteza cerebral, según una topografía ordenada, como es el caso de los núcleos VPL y VPM y el núcleo DM.
La segunda categoría viene dada por todos los núcleos cuyas eferencias fundamentalmente se dirigen a áreas corticales funcionalmente homogéneas, proyectándose también sobre otras áreas corticales alejadas de las primeras, aunque funcionalmente iguales. Se incluirían en esta categoría el LP y el pulvinar.
La tercera categoría engloba aquellos núcleos que envían sus eferencias a varias áreas corticales funcionalmente diferentes. Pertenecen a esta categoría los núcleos intralaminares, VL y VA. Por ejemplo hacia la corteza motora y hacia la corteza límbica.
La última categoría, incluye a los núcleos talámicos que se proyectan de manera difusa, en zonas corticales sensoriales y perisensoriales somatoauditivas. El complejo posterior y en particular por el CGMmc son los representantes de esta categoría.
Hemos de señalar también que existen proyecciones recíprocas de todos los núcleos de relevo y de algunos núcleos de asociación que van desde el tálamo a la corteza y desde la corteza al tálamo a través de la cápsula interna, denominadas radiaciones talámicas. Tales fibras irradian desde y hacia el tálamo como si fuera un abanico dando así lugar a la corona radiada. A pesar de que estas radiaciones establecen conexiones prácticamente con todas las partes de la corteza, la riqueza de las conexiones varia en las diferentes áreas corticales. Las más abundantes son hacia la circunvolución precentral y postcentral, área calcarina circunvolución de Heschl, región parietal posterior y partes adyacentes del lóbulo temporal [2]. Se agrupan en cuatro grupos de fibras o pedúnculos talámicos:
1.- Pedúnculo anterior, asciende hacia el lóbulo frontal por el brazo anterior de la cápsula interna. Constituido por la proyecciones del núcleo anterior del tálamo con el cíngulo y del DM con la corteza prefrontal.
2.- Pedúnculo superior, finaliza en la corteza parietal tras recorrer la porción lenticulotalámica de la cápsula interna. Formado por la fibras del núcleo VP y los núcleos VA y VL.
3.- Pedúnculo inferior, ocupa la porción sublenticular de la cápsula interna y contiene la radiación auditiva que finaliza en la corteza auditiva.
4.- Pedúnculo posterior, a través de la porción retrolenticular del brazo posterior de la cápsula interna se dirige al lóbulo occipital. Lo integran las proyecciones o radiaciones ópticas del CGL y del complejo nuclear talámico lateral posterior-pulvinar.
Conexiones de los núcleos talámicos
A continuación analizaremos las principales aferencias que llegan a los diferentes núcleos talámicos y las eferencias que de ellos parten. Utilizaremos para este fin la clasificación anatomo-funcional mencionada anteriormente.
1.- Grupo nuclear anterior
Las principales aferencias a este grupo nuclear proceden del hipotálamo (cuerpos mamilares) y formación hipocámpica. El tracto mamilo-talámico está topográficamente organizado, de modo que el núcleo mamilar medial se proyecta ventralmente al AV, AM y el lateral lo hace al AD. Las fibras de la mitad lateral del tracto, que comprende todas las fibras procedentes del núcleo mamilar lateral y la mitad lateral del medial, se proyectan de forma bilateral. El resto son homolaterales.
Las principales eferencias las envía hacia la circunvolución cingulada (corteza asociativa límbica). También envía sus axones a la corteza prefrontal, corteza motora medial, áreas orbitarias de la corteza frontal y áreas de asociación visual.
2.- Núcleo dorsomedial
Las aferencias del núcleo DM proceden del bulbo olfatorio (DMmc), la amigdala (DMmc), sustancia negra (DMpl), pálido (DMmc, DMpc), TCS (DMpl), núcleos talámicos intralaminares y de la hilera dorsal.
Las conexiones del DM-córtex prefrontal y el córtex frontal-DM son extitatorias -glutaérgicas-, mientras que las aferencias procedentes del pálido y sustancia negra son inhibitorias -GABA-érgicas- [15]. El DM estable conexiones recíprocas con diversas áreas corticales donde sus fibras acaban en la capa I, III y VI. Entre ellas destacan la corteza prefrontal e insular (DMmc, DMpc), la corteza de asociación (DMmc) y la corteza del campo ocular (área 8) (DMpl).
GRUPO NUCLEAR LATERAL
ZONA DORSAL
El núcleo DL recibe impulsos del hipocampo (a través del fórnix) tal vez de los cuerpos mamilares y áreas asociativas de la corteza parietal. Se proyecta al giro del cíngulo y zonas asociativas del lóbulo parietal.
No se conocen bien las aferencias del LP, pero parece ser que proceden de los núcleos de relevo adyacentes en especial del núcleo VP, CGL del TCS y de la corteza sensorial secundaria (área 2) con la que establece conexiones recíprocas. Los estudios de transporte retrógrado indican que el LP emite sus eferencias a las áreas asociativas 5 y 7 de la corteza parietal [2]. Se proyecta también a la corteza visual (áreas 17, 18 y 19).
Zona ventral
El núcleo VA recibe aferencias fundamentalmente del núcleo dentado del cerebelo, del globo pálido y de las porciones reticulares de la sustancia negra y áreas no motoras como el TCS. Hasta él llegan también fibras procedentes de los núcleos intralaminares y de la línea media. Las eferencias se dirigen fundamentalmente hacia los núcleos intralaminares, áreas 6 y 8 y áreas orbitofrontales.
Las aferencias del núcleo VL proceden fundamentalmente de los núcleos cerebelos profundos. El sistema dentotalámico constituye la principal aferencia. Aunque el sistema de fibras pálido-talámicas se proyectan en su mayor parte sobre las neuronas del núcleo VA algunas fibras alcanzan el núcleo VL. Hasta esta zona talámica llegan también aferencias procedentes de la corteza prefrontal, motora primaria (área 4) y núcleos intralaminares.
Este núcleo envía sus eferencias hacia la corteza motora primaria, premotora y motora suplementaria y áreas somatosensitivas no primarias en la corteza parietal (áreas 5 y 7).
Los núcleos cerebelosos profundos se proyectan fundamentalmente al VL y desde el pálido preferentemente al VA.
El núcleo VP constituye el principal destino de las fibras del lemnisco medio, lemnisco trigeminal y el tracto espino-talámico.
La parte oral del núcleo VPLo recibe proyecciones de los núcleos cerebelosos profundos contralaterales y se proyecta a la corteza motora primaria. La parte caudalis, VPLc recibe aferencias somatosensoriales de la médula espinal y núcleos del bulbo raquídeo a través del lemnisco medial y haces espinotalámicos.
El VPM recibe aferencias somáticas de receptores de la cara y estructuras intraorales. Las fibras trigeminales ascendentes llegan hasta este núcleo.
Ambos núcleos, VPL y VPM también reciben aferencias de la corteza somatosensorial primaria. Sus eferencias poseen una precisa proyección tópica a la corteza de la circunvolución postcentral, áreas 3, 1 y 2 (hómunculo sensitivo).
4.- Grupo nuclear posterior
El pulvinar no recibe proyecciones de las vías sensitivas largas ascendentes, excepto la parte inferior de este núcleo a la cual llega una proyección de las capas superficiales de los TCS.
La parte inferior de este núcleo y la porción adyacente del pulvinar lateral mantiene conexiones recíprocas con la corteza occipital incluida la corteza estriada. Las proyecciones del pulvinar inferior a las áreas 17,18 y 19 constituyen el enlace final en una vía visual extrageniculada.
La parte lateral del pulvinar se proyecta a la corteza temporal y recibe proyecciones recíprocas de la misma región. La parte medial se proyecta a la circunvolución temporal superior.
El complejo pulvinar-lateral posterior y el núcleo DM se conocen en conjunto como núcleos talámicos multimodales de asociación. Se pueden considerar conjuntamente tanto desde el punto de vista de sus conexiones como de sus funciones. Reciben proyecciones desde la retina y TCS, se relacionan con la corteza P-T-O asociativa y también con áreas visuales de la corteza cerebral. Desde un punto de vista funcional se le relaciona con la vía visual y también con el control de movimientos oculares.
La parte ventral del CGM recibe las proyecciones del núcleo central del tubérculo cuadrigémino inferior (TCI) y sus eferencias se dirigen hacia la corteza auditiva primaria (área 41). Esta proyección está tonotópicamente organizada de manera que las frecuencias bajas se sitúan lateralmente y las frecuencias altas lo hacen medialmente. A la región dorsal, llegan aferencias fundamentalmente procedentes del núcleo pericentral del TCI, su organización tonotópica no es tan clara como la de la región ventral y se proyecta a la corteza auditiva secundaria (áreas 22 y 42). La región medial recibe las proyecciones del núcleo externo del TCI y se proyecta sin tonotopía a toda la corteza auditiva (áreas 22, 41 y 42).
El CGL recibe proyecciones de la retina por la cintílla óptica y se proyecta a la corteza calcarina (área 17) a través del haz geniculocalcarino o radiación visual y recibe fibras corticogeniculadas del mismo área. En menor medida se proyecta a las áreas asociativas visuales adyacentes. Establece conexiones internucleares con el pulvinar. Las proyecciones retinogeniculadas estan tópicamente organizadas. Los cuadrantes superiores de ambas retinas (campo visual inferior) finalizan en la mitad superomedial del CGL. Los cuadrantes inferiores (campo visual superior) lo hacen en la mitad inferolateral del núcleo.
NÚCLEOS DE LA LÍNEA MEDIA
Las principales aferencias hacia estos núcleos proceden del hipotálamo, núcleos del tronco encefálico, amigdala y giro parahipocampal. Envían sus proyecciones hacia la corteza límbica y el núcleo estriado ventral.
NÚCLEOS INTRALAMINARES
Se consideran clásicamente y en conjunto como núcleos que reciben y envían proyecciones de modo inespecífico o difuso. No obstante también es cierto que diferentes porciones de los mismos reciben aferencias distintas y cada una de ellas proyecta de modo topográfico preciso a los ganglios basales y a zonas concretas de la corteza cerebral, no siendo tan inespecíficos como se creía [2].
Las principales aferencias hacia estos núcleos proceden de la formación reticular. El sistema dentorrubrotalámico (cerebelo) se proyecta fundamentalmente al VL y colaterales de este sistema se proyectan a los núcleos intralaminares. La proyección principal de las fibras palidotalámicas es el núcleo VA, colatarerales de esta proyección alcanzan los núcleos intralaminares. La mayor parte de las fibras del tracto espinotalámico y lemnisco trigeminal (fibras ascendentes de las vías del dolor) se proyectan sobre el VP, pero también a los núcleos intralaminares.
Estos núcleos reciben proyecciones también de fibras corticales procedentes de las áreas motoras y premotora. Las fibras que se originan en la corteza motora (área 4) terminan en las neuronas de los núcleos centromediano, paracentral y centrolateral. Las originadas en la corteza premotora (área 6) concluyen en los núcleos parafascicular y centrolateral. En contraste con otros núcleos talámicos, las conexiones entre los núcleos intralaminares y la corteza cerebral no son recíprocas.
Las principales eferencias de los núcleos intralaminares se dirigen a otros núcleos talámicos (fundamentalmente reticulares), estriado (caudado y putamen) y ampliamente sobre áreas asociativas de la corteza cerebral. Por métodos de marcaje retrógrado se ha visto que un pequeño número de neuronas intralaminares, proyectan por colaterales axónicos en áreas corticales bastantes alejadas entre si [16].
7.- Núcleos reticulares
Recibe proyecciones cortico-talámicas, pero carece de proyecciones tálamo-corticales. Se encuentran conectados con muchos núcleos talámicos y además son atravesados, recibiendo colaterales, de las fibras tálamo-corticales y cortico-talámicas, ejerciendo por tanto una acción moduladora sobre la actividad neuronal talámica.
Incidencia del tálamo en los procesos psicofuncionales básicos: sensitivo/motor
El tálamo junto con la corteza cerebral juegan un papel importante en el análisis e integración de las funciones sensitivas. Toda la información sensorial excepto la olfativa (la información se transmite directamente a la corteza temporal medial) se dirigen al tálamo donde hacen escala y se proyectan a las correspondientes áreas corticales específicas.
El CGM está relacionado con la vía auditiva. El input es bilateral, aunque predominan las aferencias del oido opuesto. Las aferencias de este núcleo, se dirigen hacia las áreas auditivas 41 y 42 (áreas auditivas primaria y secundaria auditiva, respectivamente) y hacia el complejo talámico asociativo dorso-pulvinar, de cual salen eferencias hacia las áreas 21 (área inferotemporal visual, circunvolución temporal, relacionada con la visión de la forma) y 22 (corteza auditiva superior. Área de Wernicke) de la corteza cerebral.
El tálamo está implicado también en los mecanismos de la visión. Las aferencias procedentes de la retina terminan en el CGL. Las eferencias se dirigen hacia la corteza visual (área 17) y hacia el complejo asociativo dorso-pulvinar para proyectarse hacia las áreas 18 (corteza visual primaria), 19 (visual secundaría), 1b (somatosensorial primaria), 39 (asociativa parieto-temporo occipital) y 37 (asociativa parieto-temporo-occipital) de la corteza cerebral.
El tálamo forma parte del sistema somatosensitivo colaborando en la percepción de estímulos mecánicos, térmicos y dolorosos. El núcleo VP recibe los tractos ascendentes largos que conducen las modalidades sensoriales, incluso del gusto, de la mitad contralateral del cuerpo y la cara. Este núcleo envía eferencias al pulvinar y al núcleo LP. Estos a su vez envían eferencias a la corteza parietal zonas esta relacionadas con el reconocimiento somatoestésico.
Gracias a las proyecciones del VP hacia las áreas 5 (corteza sensorial somestésica terciaría, área asociativa parietal posterior), 7 (áreas asociativa parietal posterior, relacionada con la percepción visuo-motora) y área 40 (asociativa parieto-temporo-occipital) es posible llevar a cabo funciones como es el reconocimiento de los objetos por el tacto (esterognosia) y del propio cuerpo (somatognosia).
El VPL actúa como relevo para la información somática del cuerpo y las extremidades, ya que dirige sus proyecciones hacia la corteza somestésica primaria en la circunvolución postcentral (área 3, 1, 2,) en la cual se analiza la información sensitiva cutánea, muscular, tendinosa, articular y visceral, siendo posible de esta manera las percepciones objetivas como la forma, el tamaño, la textura, la temperatura y el peso.
El VPM sirve de centro de relevo sensitivo-talámico de la cabeza y cara. Las eferencias de este núcleo se dirigen a través de la cápsula interna hasta la corteza somestésica primaria del lóbulo parietal.
A través de la proyecciones de esta zona talámica hacia zonas frontales (áreas 4, 8, 6, 44 y 45) el tálamo está involucrado en la sensopercepción de los movimientos.
El tálamo está implicado también en los mecanismos del dolor. Los principales núcleos de destino de los axones ascendentes para el dolor y la temperatura, se encuentran el núcleo VP. El VPM y VPL reciben la mayor parte de estas aferencias. El VPM reciben información nociceptiva desde la cara y el VPL del resto del cuerpo. La disposición similar de los estímulos mecanosensitivos y nocivos es la responsable de los mecanismos discriminativos del dolor [19].
Los núcleos talámicos intralaminares en cuanto al dolor se refiere participan en la evocación de la respuesta desencadenada por un estímulo nocivo a través de las proyecciones que llegan a estos núcleos desde la formación reticular.
Algunas modalidades sensitivas se perciben a nivel talámico, hecho este que se pone de manifiesto cuando existen lesiones o ablaciones de la corteza cerebral. En estos casos tras la lesión se pierde toda la sensibilidad contralateral a la lesión recuperándose el dolor, la temperatura y la sensibilidad epicrítica (burda). En la clínica está bien descrito este cuadro, conocido como síndrome talámico. En estos casos el umbral de estimulación que producen estas sensaciones es elevado, las modalidades sensoriales son exageradas y displacenteras, además se suelen acompañar con una marcada respuesta afectiva, normalmente atribuible a la indemnidad del núcleo DM (frecuente en lesiones vasculares).
Lesiones vasculares que afectan al territorio talámico postero-lateral (núcleos VPL, VPM, CGM, pulvinar y centromediano) pueden dar lugar a pérdida sensorial contralateral, paréstesias y dolor talámico. Bien descrito es el síndrome de Dejerine y Roussy, caracterizado por un dolor intenso, persistente y paroxístico a menudo intolerable, que se suele presentar en el momento de la lesión o después de un periodo de hemiparesia transitoria, hemiataxia y pérdida sensitiva hemicorporal.
Por otro lado la participación del tálamo en el control motor queda reflejado por las aferencias procedentes de núcleos grises de la base, cerebelo y corteza motora que llegan a él y las eferencias que de él parte hacia la corteza motora y premotora. En el sistema motor intervendran fundamentalmente los siguientes núcleos: VA y VL, núcleos intralaminares y núcleos reticulares y podemos destacar dos grandes sistemas: palidal y cerebeloso. La separación entre ambos circuitos es debido a que las aferencias son distintas y también sus eferencias hacia áreas corticales en donde proyectan. Alteraciones en las proyecciones del VL pueden dar lugar a trastornos motores (discinesias). Lesiones en este núcleo disminuyen los movimientos anormales cerebelosos y de los núcleos grises de la base [6].
Lesiones en el núcleo ventral intermedio (Vim), núcleos ventrales caudales, centromediano, núcleos sensoriales y pulvinar pueden causar gran variedad de alteraciones del movimiento, incluyendo, distonias, temblor, balismo, corea, entre otros [20-22]. Lesiones vasculares que afectan a los núcleos VA, VL, DM y núcleo anterior pueden causar hemiparesia contralateral y trastornos de los campos visuales.
Existe evidencia de que los núcleos intralaminares también están implicados en el control de los movimientos. Este núcleo recibe aferencias principalmente de la formación reticular, del pálido, putamen, núcleos subtálamicos y de áreas corticales (6 y 4). Las conexiones que estos núcleos mantienen con el putamen y el caudado, contribuyen al control motor subcortical.
El núcleo centromediano recibe aferencias del pálido, sustancia negra (zona reticular), zona incierta, núcleos profundos del cerebelo, córtex motor primario y núcleos reticulares [23,24]. Envía amplias proyecciones glutamato-érgicas excitatorias al putamen y proyecciones difusas al borde dorsolateral del núcleo caudado y núcleos subtalámicos [25, 26]. Los núcleos reticulares talámicos, terminan de manera difusa en la corteza cerebral, permitiendo la activación necesaria para el correcto funcionamiento del sistema motor.
Existen trabajos que señalan cierta implicación de los núcleos de la línea media con el sistema motor. Lee y Marsden [21] que señalan las lesiones causantes de las distonías tálamicas no hay que situarlas en los núcleos VA y VL sino en zonas más posteriores o en los núcleos de la línea media.
Podemos describir una semiología motriz que caracterizaría a las lesiones talámicas: 1.- alteraciones del sistema motor voluntario (incoordinación cerebelosa contralateral, sincinesias homolaterales de imitación y contracturas); 2.- alteraciones del sistema motor involuntario; 3- perturbaciones globales del movimiento (mano talámica, caracterizada por movimientos incesantes de los dedos, tanto en el plano horizontal como en el vertical); 4.- alteraciones de la marcha [27].
Incidencia del tálamo en los procesos psicofuncionales superiores: nivel atencional/ emoción/ lenguaje/ memoria/ función ejecutiva
El tálamo regula funciones de la corteza asociativa y es importante en funciones como el lenguaje, el habla y funciones cognitivas, mediadas corticalmente [28].
Hay tres regiones importantes de corteza asociativa: parieto-temporo-occipital, prefontral y límbica, hacia las cuales se proyectan diferentes núcleos talámicos. Así la corteza parieto-temporo-occipital (áreas 39 y 40) está relacionada con funciones perceptivas, visión, lectura y recibe información del pulvinar.
La corteza asociativa prefrontal es importante para la planificación de la conducta y los movimientos, cognición, aprendizaje, memoria y pensamiento. El DM proyecta sus fibras hacia esta zona cortical. Un estudio reciente realizado en monos a los cuales se les realizó una ablación del núcleo DMmc, ha puesto de manifiesto que lesiones en esta zona talámica causan trastornos de memoria debidos principalmente a la interrupción de la función entre este núcleo y el cortex prefrontal [29].
La corteza límbica, relacionada con el aprendizaje, la memoria y la emoción, recibe fundamentalmente eferencias del núcleo anterior talámico.
Tálamo y nivel atencional
La participación del tálamo y de la formación reticular en la regulación del nivel de arousal (vigilancia) se puso de manifiesto ya en la primera mitad del siglo XX con los trabajos pioneros realizados por Morison y Dempsey [30], Jasper [31] y Moruzzi y Magoun [32].
Los núcleos intralaminares están relacionados con la excitabilidad general de la corteza cerebral al transmitir información procedente de la formación reticular mesencefálica a múltiples áreas corticales y al cuerpo estriado, desempeñando un papel importante en el control del sueño y la vigilia. La estimulación eléctrica de estos núcleos provoca una activación generalizada de la corteza cerebral (recruiting response) formando parte del sustrato anatómico del sistema reticular activador ascendente y por lo tanto de los mecanismos del sueño y la vigilia.
Los núcleos de la línea media parecen ser el lugar por el que el tálamo, junto con la formación reticular, controla las señales que acceden a la corteza cerebral. Trabajos realizados en este campo, señalan que el tálamo regula el nivel de arousal cortical a través de las conexiones tálamo-corticales que se originan en los núcleos DM, intralaminares y de la línea media y a través de las interacciones intratalámicas con los núcleos reticulares [23,33].
Los estudios llevados a cabo en especies animales han proporcionado evidencias de que los núcleos reticulares están relacionados con el ciclo sueño-vigilia [23,33]. Se ha comprobado que las neuronas GABA-érgicas de los núcleos reticulares controlan la actividad de las neuronas tálamo-corticales, modulando así la actividad cortical [33,34].
En humanos se ha visto en estudios realizados con técnicas de neuroimagen funcional que existen variaciones en el flujo sanguíneo tálamico en función del nivel de conciencia [35,36]. Kinomura, Larsson, Gulyás y Roland [37] han demostrado cambios en el flujo sanguíneo de los núcleos intralaminares del tálamo y la formación reticular en función del nivel de arousal de sujeto.
En una investigación llevada a cabo por Fiset y cols. [38] donde se manipulaba el nivel de conciencia de los sujetos utilizando Propofol (fármaco con propiedades anestésicas que disminuye el flujo sanguíneo cerebral, lo cual se acompaña de una reducción del requerimiento metabólico cerebral de oxígeno y de la disminución de la presión intracraneal), encontraron relación negativa entre el flujo sanguíneo talámico (con PET) y la concentración de propofol utilizada. Los efectos de esta droga anestésica son más pronunciados en la zona medial talámica, el giro cingulado, giro orbitofrontal y giro angular. Parece ser que las variaciones observadas en el tálamo (especialmente en la zona medial) están significativamente relacionadas con la actividad de la formación reticular. Estos autores sugieren que el sistema reticulo-talámico juega un papel fundamental en la modulación de la conciencia.
En la clínica se ha observado que lesiones vasculares en los núcleos intralaminares y dorsomediales pueden causar mutismo acinético y el síndrome de Kleine-Levin (síndrome de hipersomnia y bulimia). Este síndrome se caracteriza por periodos recurrentes de excesiva somnolencia, hiperfagia, hipersexualidad y alteraciones de la memoria reciente.
Diferentes aspectos de la atención pueden ser atribuibles al córtex prelímbico y núcleo DM [39]. Infartos tálamicos pueden causar negligencia y déficits atencionales del espacio extrapersonal contralateral a la lesión [40-42].
Tálamo y emoción
El tálamo interviene en los procesos emocionales y motivacionales. Los principales núcleos implicados son el VA, DM y grupo nuclear anterior.
El VA recibe aferencias desde el cuerpo mamilar y proyecta fibras hacia el cíngulo.
El núcleo DM recibe desde el hipotálamo y la amigdala, enviando sus fibras hacia el lóbulo prefrontal. El DM con sus proyecciones hasta la corteza prefrontal y estructuras límbicas, participa en la integración de la información visceral con el afecto, las emociones y el pensamiento.
El grupo anterior media información visual y emocional. La estimulación eléctrica y la ablación de este núcleo inducen cambios en la tesión arterial y los impulsos motivacionales.
Tálamo y lenguaje
Penfield y Roberts en [43] fueron los primeros en destacar que el tálamo con sus extensas proyecciones corticales está relacionado con funciones linguísticas.
En el lenguaje intervienen fundamentalmente el pulvinar, el grupo nuclear lateral (fundamentalmente el VPL y VPM) y el grupo nuclear anterior. Existen conexiones reciprocas entre el pulvinar y la corteza cerebral importantes para el lenguaje y el pensamiento simbólico (hacia la encrucijada funcional parieto-temporo-occipital). El VPL y VPM participan en el lenguaje gracias a las relaciones que mantienen con áreas somestésicas y a la integración específica que en ellos se produce.
Existe evidencia electrofisiológica de la participación del tálamo en los aspectos motores del lenguaje. Mateer [44] encontró un incremento en la duración de la respuesta verbal después de estimular el tálamo izquierdo dando como resultado una mala pronunciación de las palabras y cambios articulatorios. Posteriormente Bhatnagar y Andy [45] observaron tras la estimulación del núcleo centromediano izquierdo espasmos motores articulatorios.
Johnson y Ojemann [46] señalan que la zona ventro-lateral del tálamo izquierdo (especialmente la parte central) participa en la integración de los mecanismos motores del habla, incluyendo la respiración, ya que tras la estimulación de esta zona talámica se observa una inhibición de la respiración, enlentecimiento del habla y presencia de perseveraciones.
El pulvinar, no está sólo intercalado entre las vías óptica y acústica, sino que proyecta a zonas corticales importantes para el lenguaje y el pensamiento simbólico (encrucijada parieto-temporo-occipital). Lesiones en el núcleo anterior o en el pulvinar pueden causar anomia, parafasias semánticas y errores sintácticos [47].
Ojemann [48] encontró que tras la estimulación de la zona anterior (parte más lateral) del tálamo aparecen repeticiones de palabras que previamente han sido denominadas correctamente. Si la estimulación se realiza en la parte central de la zona ventrolateral aparecían perseveraciones. La estimulación de la parte posterior de la zona ventrolateral y pulvinar anterior daba lugar a la aparición de omisiones y errores en la denominación de objetos.
Tálamo y memoria
En cuanto a la memoria se refiere parece ser que son los núcleos talámicos anteriores, núcleos de la línea media, núcleos dorsomediales y núcleos intralaminares los implicados en los procesos mnésicos, aunque no existen evidencias concluyentes que indiquen cual de estas estructuras es crucial para el buen funcionamiento de la memoria anterógrada [49].
Weiskrantz [50] señala que los déficits de memoria que suelen aparecer en pacientes con lesiones talámicas son similares a los observados tras lesiones en el lóbulo temporal medial: déficits en la codificación de nueva información dando como resultado una alteración en la memoria anterógrada, estando intacta la memoria a corto plazo.
Existe evidencia de alteraciones mnésicas tras lesiones talámicas específicas, en especial en el núcleo DM [51], núcleo anterior [52, 53] y núcleos intralaminares [54].
Parece ser que el núcleo anterior está relacionado con el proceso de consolidación de la información permitiendo la formación de trazos mnésicos y con la memoria de trabajo [55].
Recientemente Celerier, Ognard, Decorte y Beracochea [56] han demostrado en ratones que lesiones en el núcleo anterior causan alteraciones en la ejecución de tareas mnésicas. Según estos autores este grupo nuclear está relacionado con el mantenimiento de la información en el tiempo independientemente de la naturaleza de la información y con los procesos asociativos de la información unimodal y polimodal.
Los núcleos anteriores del tálamo están implicados en los procesos de organización temporal de la memoria [57]. Los núcleos intralaminares permiten la salida de trazos mnésicos ya memorizados, es decir el proceso de activación.
En los procesos de organización temporal de los recuerdos recientes y antiguos intervienen los núcleos DM. Lesiones en estos núcleos pueden dar lugar a una desorganización temporal del recuerdo que afectaría no solo a la información nueva, sino también a la antigua. Pueden aparecer fabulaciones como las descritas en el síndrome de Korsakoff. Victor, Adams y Collins [58] que en los pacientes con síndrome de Korsakoff el núcleo DM está afectado en el 100% de los sujetos, junto con los cuerpos mamilares. El déficit es más severo si están implicados los núcleos DM del tálamo y los núcleos de la línea media [59]. Además, en el síndrome de Korsakoff [60] se ha encontrado relación entre la amnesia anterógrada y el grado de atrofia en los núcleos de la línea media, no evidenciándose relación con la atrofia en los cuerpos mamilares, hipocampo o giro parahipocampal.
Gaffan y Parker [29] en un estudio realizado con monos han encontrado que la parte magnocelular del núcleo DM juega un papel importante en la memoria. Una lesión en esta zona da lugar a una alteración en esta función cognitiva atribuible a la desconexión con el córtex prefrontal.
Sin embargo y a pesar de estos resultados, todavía existe controversia de si lesiones en el DM pueden causar déficits de memoria. En una extensa revisión realizada por Van der Werf y cols [61] sobre los déficits neuropsicológicos que pueden aparecer tras infartos talámicos señalan, que no existe evidencia suficiente para poder establecer la relación del DM con los problemas mnésicos que ocurren después de lesiones diencefálicas. Concluyen que los déficits mnésicos que pueden aparecer y que son compatibles con un “síndrome amnésico”, dependen de la integridad del tracto mamilo-talámico.
La participación del tálamo en el procesamiento mnésico se ha puesto también de manifiesto a través de los estudios electrofisiológicos realizados. Ojemann [48] encontró que la estimulación ventrolateral talámica afecta a la memoria verbal a corto plazo. La estimulación de esta zona durante la presentación del material que posteriormente será evocado reduce el número de errores. La estimulación del pulvinar izquierdo altera el procesamiento mnésico verbal, mientras que la estimulación del pulvinar derecho altera el procesamiento mnésico no verbal [46].
Tálamo y función ejecutiva
Lesiones en el tálamo también pueden causar alteraciones en las funciones ejecutivas, atención, iniciativa, inhibición y organización temporal de la conducta, funciones estas relacionadas con el córtex prefrontal.
Se ha propuesto que entre los núcleos talámicos implicados en la función ejecutiva se encuentran el DM, los intralaminares y los núcleos de la línea media.
Algunos pacientes muestran deterioro en el funcionamiento ejecutivo después de infartos selectivos del DM [52, 62]. Mennemeier y cols. [63] han señalado que los pacientes con lesiones talámicas pueden presentar dificultad para utilizar estrategias mnésicas, más que padecer un defecto de codificación de la información. Se ha propuesto que una interrupción entre el núcleo DM y el córtex prefrontal puede ser la responsable de la aparición de estos déficits.
Sin embargo, existen datos que ponen de manifiesto la aparición de un deterioro similar en la función ejecutiva después de infartos talámicos que no implican al núcleo DM. Se han descrito como lesiones en los núcleos intralaminares y partes adyacentes de los núcleos de la línea media pueden causar déficits en la función ejecutiva [52, 63].
Van der Werf y cols. [61] señalan que lesiones que impliquen a un único núcleo tálamico, por si mismas no son suficientes para que aparezca deterioro en la función ejecutiva,siendo necesaria la afectación de dos o más núcleos (DM, intralaminares y de la línea media).
REFERENCIAS
1. Sherman SM, Guillery RW. Exploring the Thalamus. San Diego: Academic Press; 2001
2. Carpenter MB. Neuroanatomía. Fundamentos. (4ªed.). Buenos Aires: Editorial Médica Panamericana; 1994
3. Ralston HJ. Tálamo. En Wong-Riley MMT, ed. Secretos de las Neurociencias. Mexico: McGraw-Hill; 2001. pp. 275-81.
4. Amaral DG. Organización funcional de la percepción y el movimiento. En Kandel ER, Schwartz JH, Jessell TM, eds. Principios de Neurociencia (4ªed.). Madrid: McGraw-Hill Interamericana 2001; pp. 337-48.
5. Elliot HC. Textbook of Neuroanatomy. Philadelphia: Lippincatt Co; 1969
6. Afifi AK, Bergman RA. Neuroanatomía Funcional. Texto y Atlas. México: McGraw-Hill Interamericana; 1999
7. Armengol JA. Centro y vías nerviosas I. Médula espinal, tronco del encéfalo y cerebelo. En Delgado JM, Ferrús A, Mora F, Rubia FJ, eds. Manual de Neurociencia. Madrid: Síntesis; 1998. pp. 359-392.
8. Morel A, Magnin M, Jeanmonod D. Multiarchitectonic and Stereotactic Atlas of the Human Thalamus. The Journal of Comparative Neurology 1997, 387: 588-630.
9. Hirai T, Jones, EG. A new parcellation of the human thalamus on the basis of histochemical staining. Brain Research Reviews 1989, 14: 1-34.
10. Martin JH. Neuroanatomía (2ªed.). Madrid: Prentice Hall; 1997
11. Steriade M, Deschenes M. The thalamus as a neuronal oscillator. Brain Research Reviews 1984, 8: 1-63.
12. Lorente de Nó R. Cerebral cortex: Architecture, intracortical connections, motor projections. En Fulton J, ed. Physiology of the Nervous System. Oxford: Oxford University Press; 1938. pp. 291-325.
13. Macchi, G. The intralaminar system revisited. En Minciacchi D, Molinari M, Macchi MG, Jones EG eds. Thalamic Networks for Relay and Modulation. Oxford: Pergamon Press; 1993. pp. 175-184.
14. Jones EG. Viewpoint: the core and matrix of thalamic organization. Neuroscience 1998; 85: 331-45.
15. Kuroda M, Yokofujita J, Murakami K. An ultrastructurd study of the neural circuit between the prefrontal cortex and the mediodorsal nucleus of the thalamus. Progress in Neurobiology 1998; 54(4): 417-58.
16. Bentivoglio M, Macchi C, Albanese A. The cortical projection of the thalamic intralaminar nuclei, as studied in cat and rat with multiple fluorescent retrograde tracing techique. Neurosci Lett 1981; 26: 5-10.
17. Stephens RB, Stilwell DL. Arteries and veins of the human brain. Springfiel: Charles C. Thomas; 1969.
18. Tatu L, Moulin Th, Bogousslavsky J, Duvernoy H. Arterial territories of the human brain. Neurology 1998; 50, 1699-1708.
19. Purves D, Augustine GJ, Fitzpatrick D, Katz LC, LaMantia AS, McNamara JO. Invitación a la Neurociencia. Madrid: Editorial Médica Panamerica; 2001
20. Ghika j, Bogousslavsky J, Maeder P y Regli F. The “Jerky distonic unsteady hand”: a delayed motor syndrome in posterior thalamic infarctions. Journal of Neurology 1994; 241: 537-42
21. Lee MS, Marsden CD. Movement disordres following lesions of the thalamus or subthalamic region. Movement Disorders 1994; 9(5): 493-507.
22. Lee MS, Kim YD, Yang JW, Lyoo CH, Oh SH, Kim HS. Clinical and anatomical factors associated with thalamic dyskinesias. Journal of the Neurological Sciences 2001; 182: 137-142.
23. Steriade M, Parent T, Hada A. Thalamic projections of nucleus reticularis thalami of cat: a study using retrograde transport of horseradish peroxidase and fluorescent tracers. J. Comp. Neurol 1984; 229: 531-47.
24. Royce GJ, Bromley S, Gracco C. Subcortical projections on the centromedian and parafascicular thalamic nuclei in the cat. J Comp Neurol 1991; 306: 129-55.
25. Parent A. Extrinsic connections of the basal ganglia. Trends in Neurosciences 1990; 13: 254-8.
26. Groenewegen HJ, Berendse HW. The specificity of the “nonspecific” midline and intralaminar thalamic nuclei. Trends in Neurosciences 1994; 17: 52-7.
27. Perea MV. Fundamentos de Neuropsicología. Psicobiología del Movimiento. Salamanca: Ediciones Universidad de Salamanca; 1989
28. Bhatnagar SC, Andy OJ. Neurociencia para el estudio de las alteraciones de la comunicación. Barcelona: Masson-Williams & Wilkins; 1997.
29. Gaffan D, Parker A. Mediodorsal thalamic function in scene memory in rhesus monkeys. Brain 2000; 123: 816-27.
30. Morison RS, Dempsey EW. A syudy of thalamocortical relations. Americam Journal of Physiology 1942; 135: 281-92.
31. Jasper HH. Diffuse projection systems: the integrative action of the thalamic reticular system. Electroencephalography and Clinical Neurophysiology 1949; 1: 405-19.
32. Moruzzi G, Magoun HW. Brain stem reticular formation and activation of the EEG. Electroencephalography and Clinical Neurophysiology 1949; 1: 455-73.
33. Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science 1993; 262: 679-85.
34. Destexhe A, Contreras D, Sejnowski TJ, Steriade M. A model os spindle rhythmicity in the isolated thalamic reticular nucleus. Journal of Neurophysiology 1994; 72: 803-18.
35. Hofle N, Paus T, Rutens D, Fiset P, Gotman J, Evans AC, Jones BE. Covariation of regional cerebral blood flow with delta and spindle activity during slow wave sleep in humans. Journal of Neurocience1997; 17: 4800-08.
36. Paus T, Zatorre RJ, Hofle N, Caramanos Z, Gotman J, Pretides M, Evans AC. Time-related changes in neural systems underlying attention and arousal during the performance of and auditory vigilance tesk. Journal of Cognitive Neuroscience 1997; 9(3): 392-408.
37. Kinomura S, Larsson J, Gulyás B, Roland PE. Activation by atttention of the human reticular formation and thalamic intralaminar nuclei. Sciencie1996; 271: 512-4.
38. Fiset P, Paus, T, Daloze Th, Plourde G, Meurte P, Bonhomme V, Hajj-Ali N, Backman SB, Evans AC. Brain mechanisms of Propofol-induced loss of consciousness in human: a Positron Emission Tomographic study. Journal of Neuroscience 1999; 19(13): 5506-13.
39. Chudasama Y, Muir JL. Visual attention in the rat: a role for the prelimbic cortex and thalamic nuclei?. Behavioral Neuroscience 2001; 115(2): 417-28.
40. Watson RT, Heilman KM. Thalamic neglect. Neurology 1979; 29: 690-4.
41. Graff-Radford NR, Damasio H, Yamada T, Eslinger PJ, Damasio AR. Nonhemorrhagic thalamic infarction. Brain; 1985; 108: 485-516.
42. Barrett AM, Schwartz RL, Crucian GP, Kim M, Heilman, KM. Attentional grasp in far extrapersonal space after thalamic infarction. Neuropsychologia 2000; 38: 778-84.
43. Penfield W, Roberts L. Speech and Brain Mechanisms. Princenton: Princenton University Press; 1959.
44. Mateer C. Asymmetric effects of thalamic stimulation on rate os speech. Neuropsychologica 1978; 16: 497-9.
45. Bhatnagar SC, Andy OJ. Allevation of acquired stuttering with human centremedian thalamic stimulation. Journal Neurology, Neurosurgery and Psychiatry 1989; 52: 1182-4.
46. Johnson M D, Ojemann GA. The role of the human thalamus in language and memory: Evidence from electrophysiological studies. Brain and Cognition 2000; 42(2): 218-30.
47. Crosson B. Subcortical functions in language in memory. New York: Guilford Press; 1992.
48. Ojemann GA. Asymmetric function of the thalamus in man. Annals of New York Academy of Science 1977; 299: 380-96.
49. Bentivoglio M, Aggleton JP, Mishkin M. The thalamus and memory formation. En Steriade M, Jones EG, McCormick DA, eds. Thalamus. Volume II. Experimental and clinical aspects. Oxford: Elservier Science Ltd; 1997; pp. 689-720.
50. Weiskrantz L. On issues and theories of the human amnesic syndrome. En Weinberger NM, McGaugh, JL, Lynch G, eds. Memory systems of the human brain: animal and human cognitive processes. New York: Guilford; 1985.
51. Guberman A, Stuss DT. The syndrome of bilateral paramedian thalamic infarction. Neurology 1983; 33: 540-6.
52. Graff-Radford NR, Tranel D, Van Hoese GW, Brandt JP. Diencephalic amnesia. Brain 1990; 113: 1-25.
53. Parkin AJ, Rees JE, Hunkin NM, Rose PE. Impairment of memory following discrete thalamic infarction. Neuropsychologia 1994; 32: 39-51.
54. Calabrese P, Haupts M, Markowitsch H, Gehlen W. Case Report: The cognitive-mnesic performance profile of a patient with bilateral asymmetrical thalamic infarction. International Journal of Neuroscience 1993; 71: 101-6.
55. Peru A, Fabbro F. Thalamic amnesia following venous infarction: evidence from a single case study. Brain and Cognition 1997; 33: 278-94.
56. Celerier A, Ognard R, Decorte L, Beracochea D. Deficits of spatial and non-spatial memory and of auditory fear conditioning following anterior thalamic lesions in mice: comparison with chronic alcohol consumption. The European Journal of Neuroscience 2000; 12(7): 2575-84.
57. Tranel D, Damasio AR. Neurobiological Foundations of Human Memory. En Baddeley AD, Wilson BA, Watts FN. eds. Handbook of Memory Disorders. Chichester: Wiley; 1995. pp. 27-47.
58. Victor M, Adams RD, Collins GH. The Wernicke-Korsakoff syndrome and related neurologic disorders due to alcoholism and malnutrition. (2nd ed.). Philadelphia: Davis; 1989.
59. Zola-Morgan S, Squire LR. Neuroanatomy of memory. Annual Review Neuroscience 1993; 16: 547-63.
60. Visser PJ, Krabbendam L, Verhey FRJ, Hofman PAM, Verhoeven WMA, Tuinier S, et al. Brain correlates of memory dysfunction in alcoholic Korsakoff’s syndrome. Journal of Neurology, Neurosurgery and Psychiatry 1999; 67(6): 774-8
61. Van der Werf YD, Witter MP, Uylings HBM, Jolles J. Neuropsychology of infarctions in the thalamus: a review. Neuropsychologia 2000; 38: 613-27.
62. Stuss DT, Guberman A, Nelson R, Larochelle S. The neuropsychology of paramediam thalamic infarction. Brain and Cognition 1988; 8: 348-78.
63. Mennemeier M, Fennell E, Valenstein E, Heilman KM. Contributions of the left intralaminar and medial thalamic nuclei to memory. Comparisons and report of a case. Archives of Neurology 1992; 49: 1050-8.
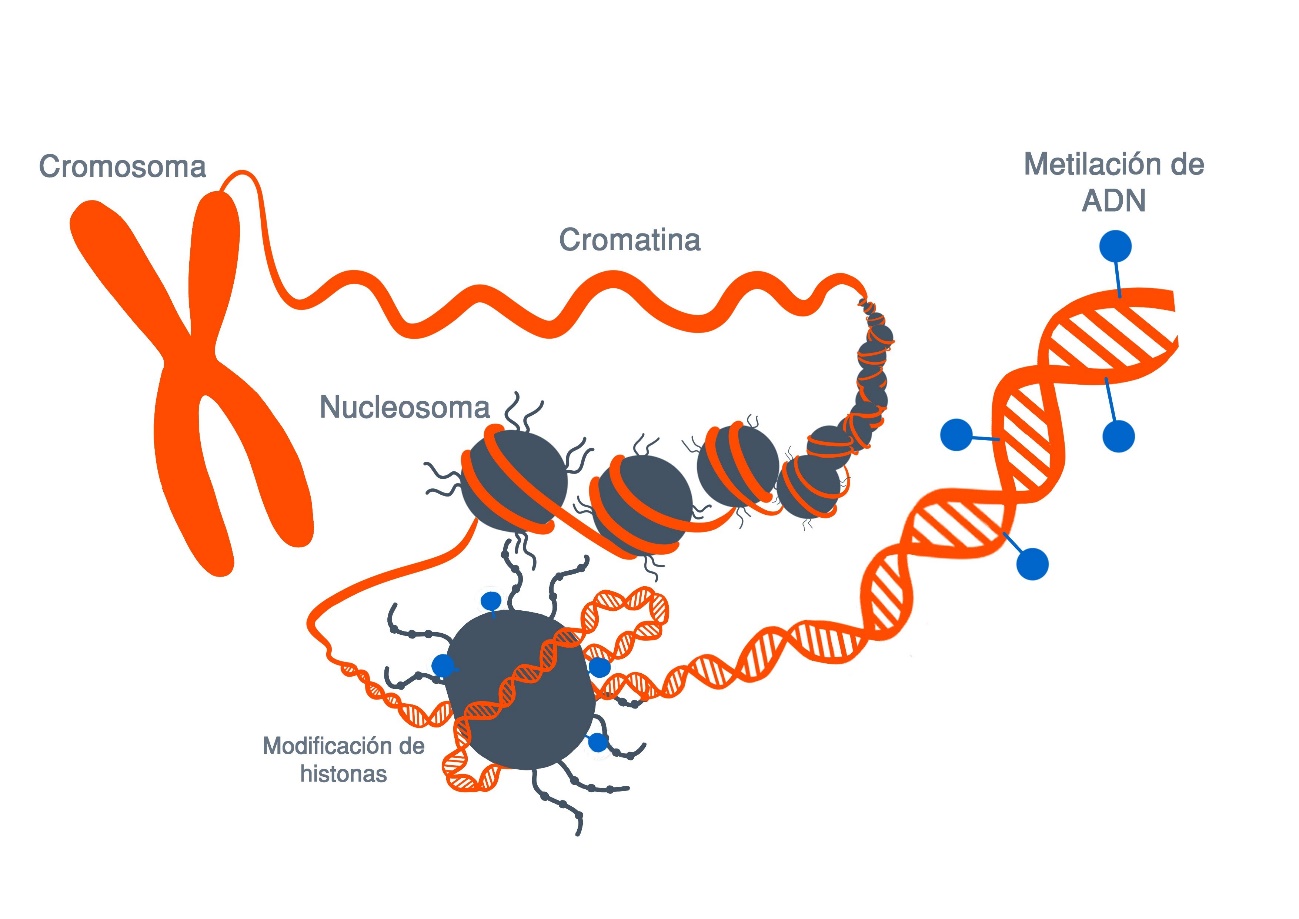
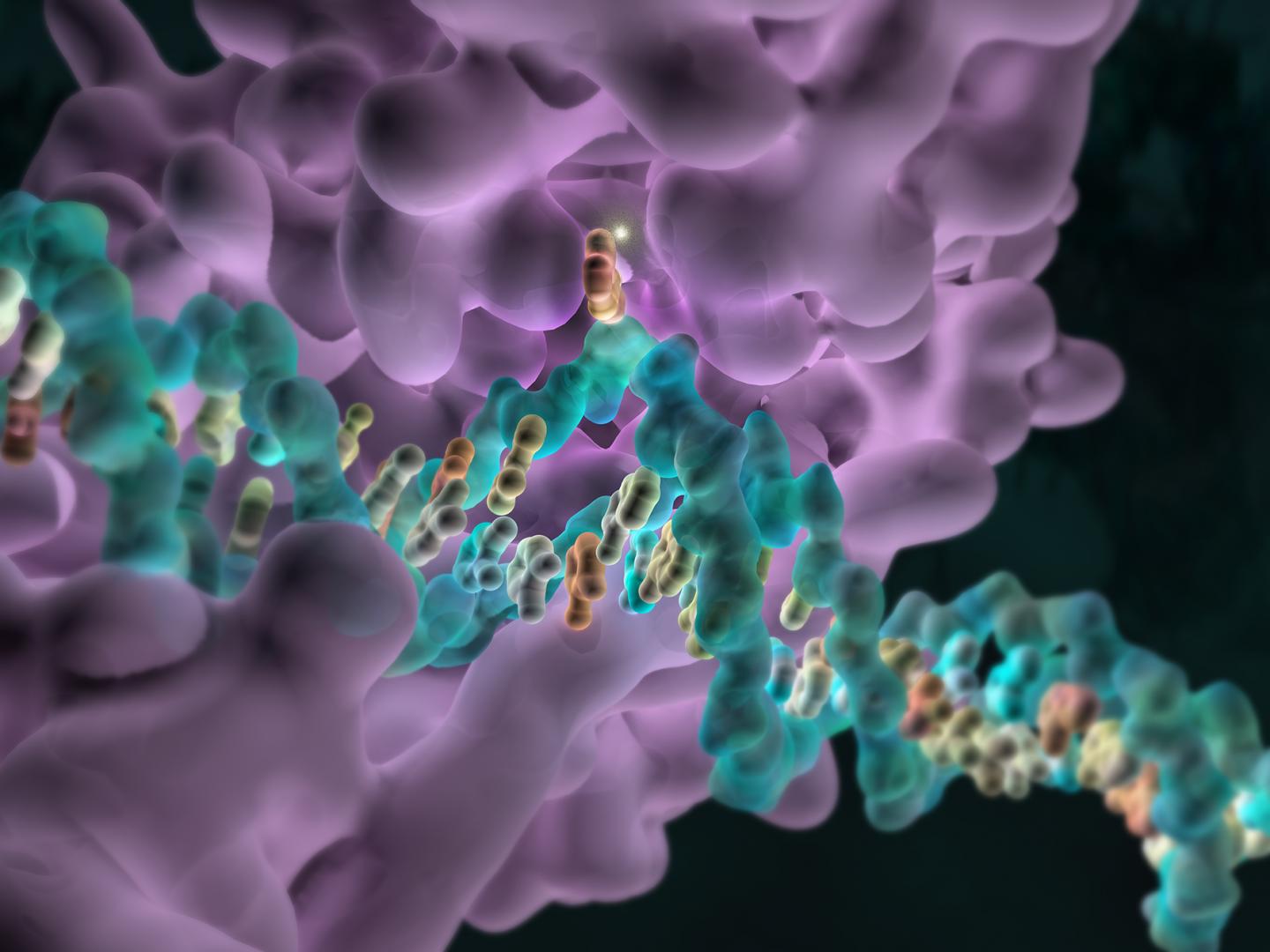 Metilacion en el dolor
Metilacion en el dolor
 Células individuales organizadas por su presencia en órganos y sistemas de todo el cuerpo humano. Foto: Aviv Regev y Anna Hupalowska, Human Cell Atlas
Células individuales organizadas por su presencia en órganos y sistemas de todo el cuerpo humano. Foto: Aviv Regev y Anna Hupalowska, Human Cell Atlas Integración computacional de perfiles de una sola célula y genética para identificar genes de enfermedades y tipos de células en todo el cuerpo humano. Foto: Aviv Regev and Anna Hupalowska, Human Cell Atlas
Integración computacional de perfiles de una sola célula y genética para identificar genes de enfermedades y tipos de células en todo el cuerpo humano. Foto: Aviv Regev and Anna Hupalowska, Human Cell Atlas

 Células individuales organizadas por su presencia en órganos y sistemas de todo el cuerpo humano. Foto: Aviv Regev y Anna Hupalowska, Human Cell Atlas
Células individuales organizadas por su presencia en órganos y sistemas de todo el cuerpo humano. Foto: Aviv Regev y Anna Hupalowska, Human Cell Atlas Integración computacional de perfiles de una sola célula y genética para identificar genes de enfermedades y tipos de células en todo el cuerpo humano. Foto: Aviv Regev and Anna Hupalowska, Human Cell Atlas
Integración computacional de perfiles de una sola célula y genética para identificar genes de enfermedades y tipos de células en todo el cuerpo humano. Foto: Aviv Regev and Anna Hupalowska, Human Cell Atlas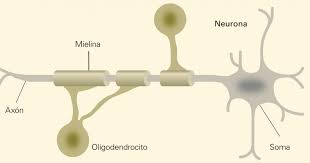

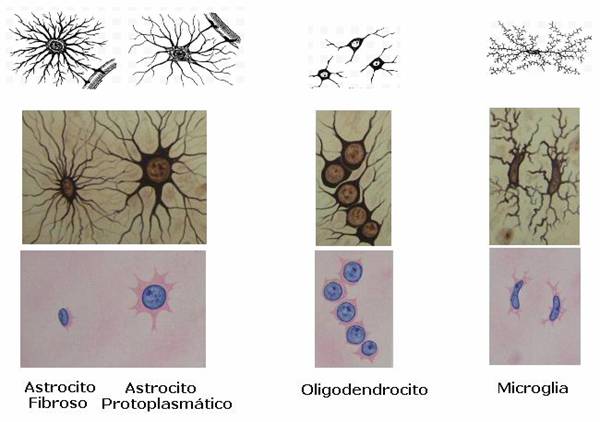 Los oligodendrocitos son un tipo de células de la
Los oligodendrocitos son un tipo de células de la 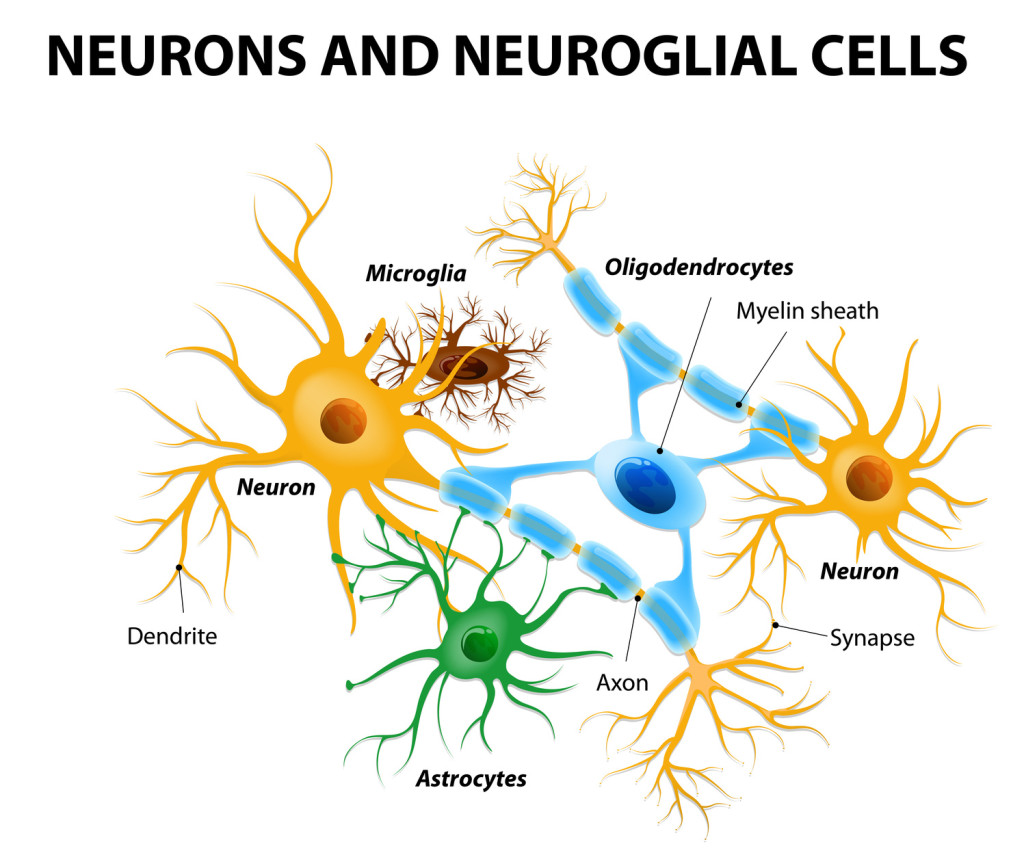


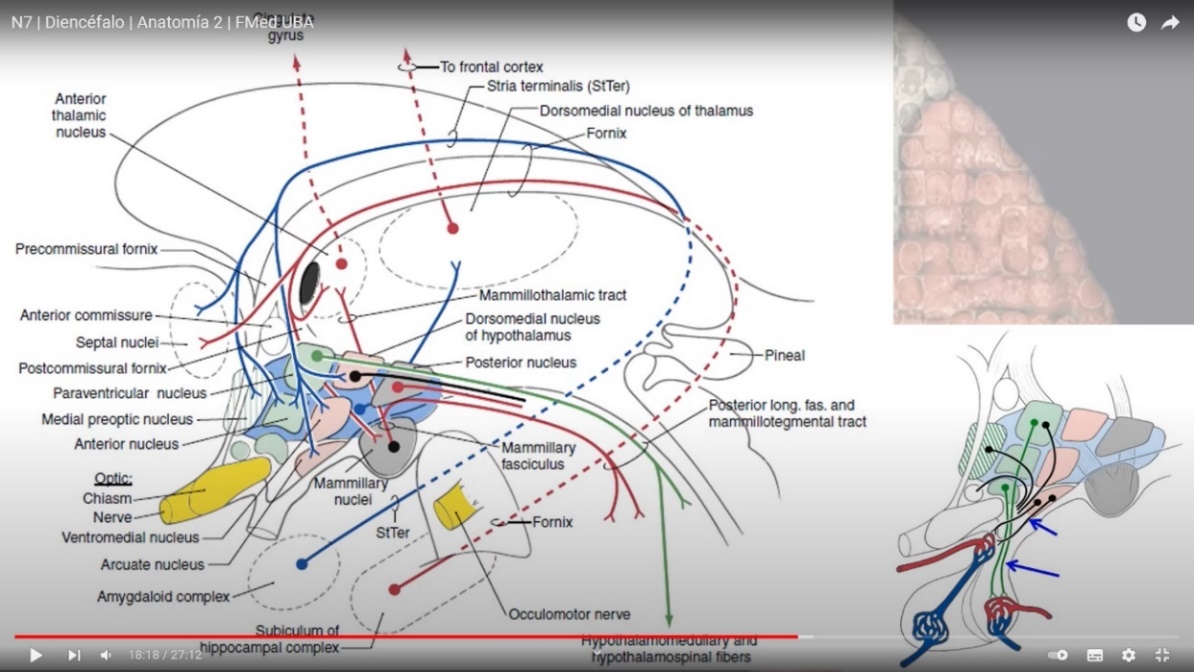 NUCLEOS DEL SEPTUM
NUCLEOS DEL SEPTUM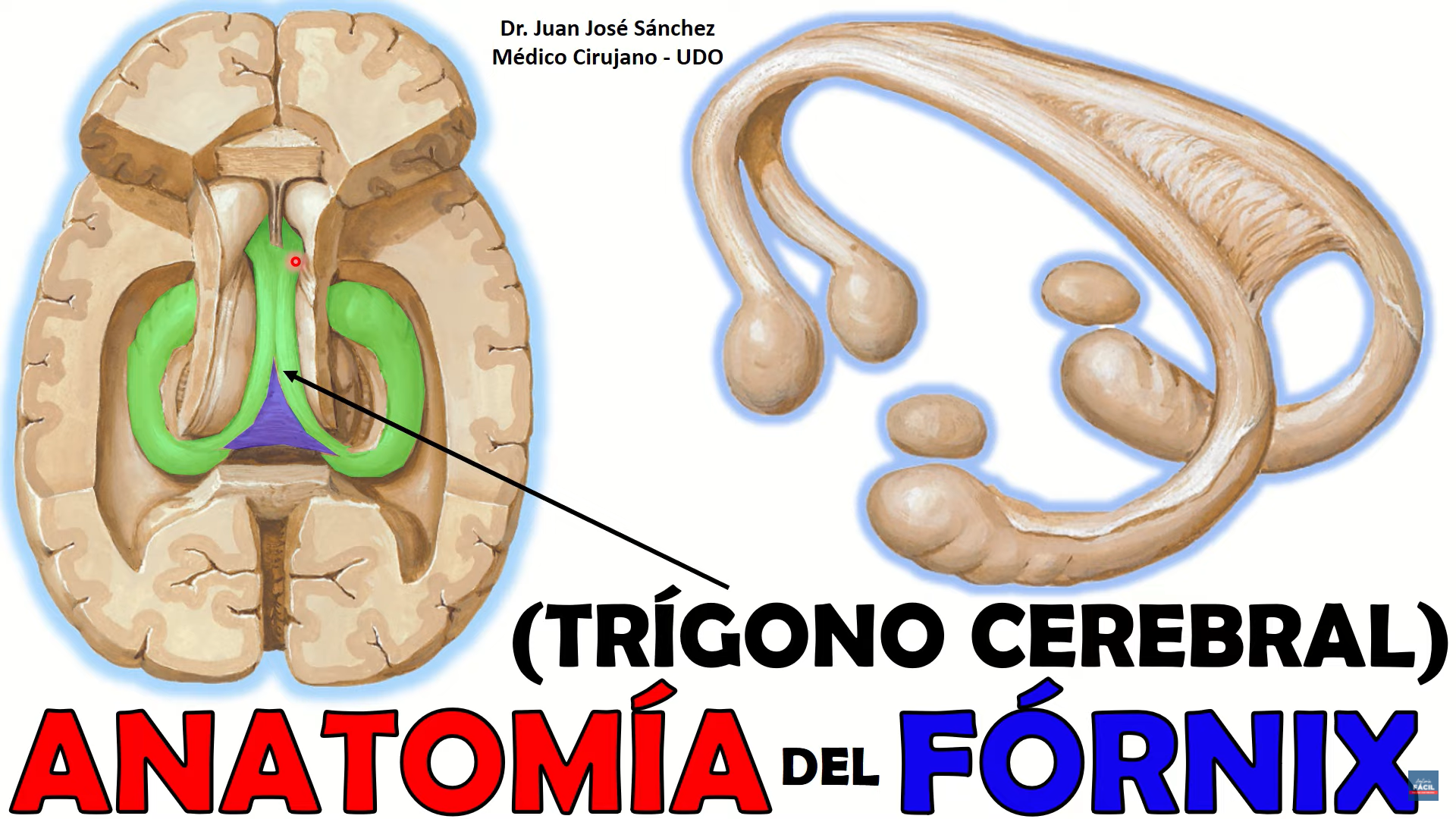
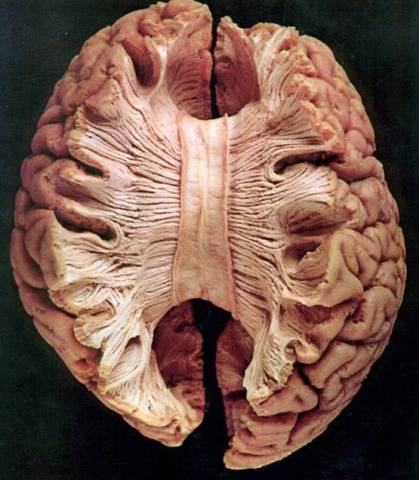
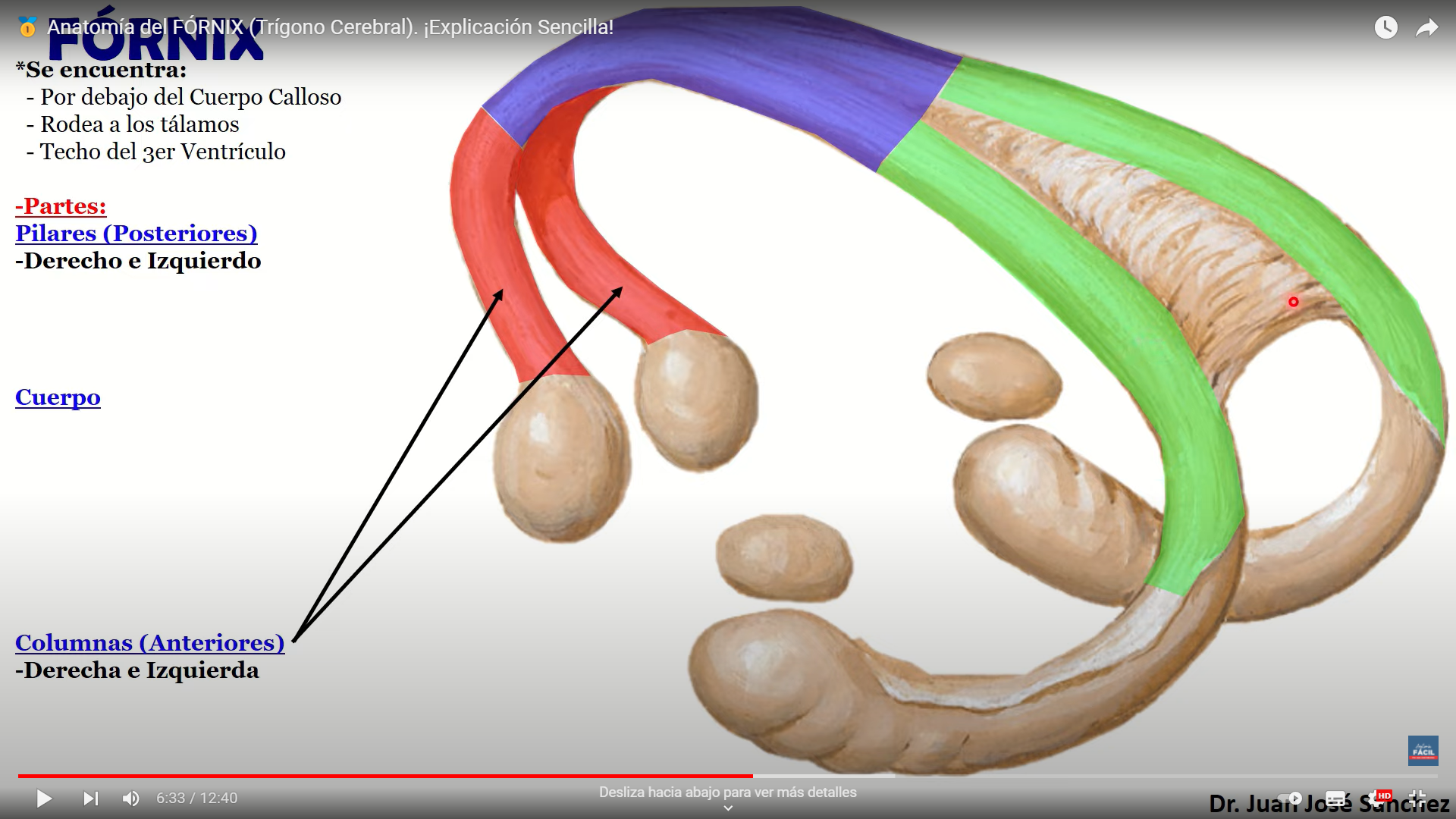
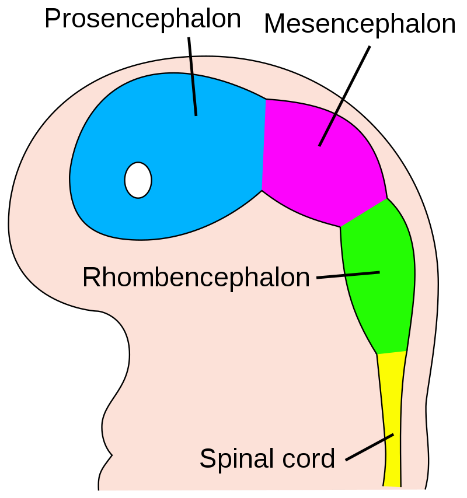
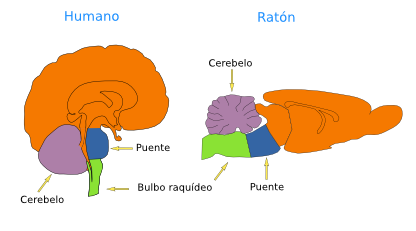
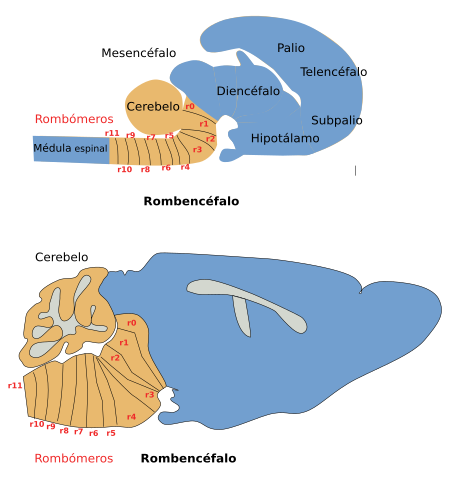
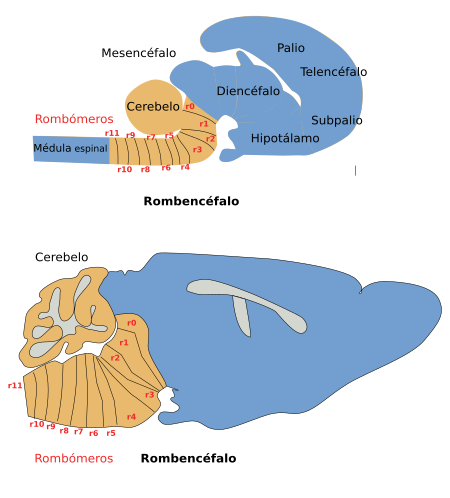
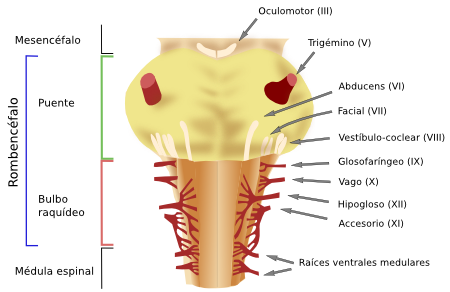 Figura 3. Localización de los nervios craneales en el rombencéfalo de un ratón. Vista ventral. (Modificado de Cordes, 2001).
Figura 3. Localización de los nervios craneales en el rombencéfalo de un ratón. Vista ventral. (Modificado de Cordes, 2001).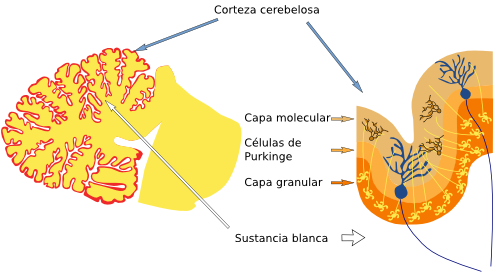 Figura 4. La imagen de la izquierda es un esquema de un cerebelo humano cortado por la línea media donde se señalan la sustancia blanca y la corteza cerebelosa (en rojo). La imagen de la derecha es un esquema de la corteza cerebelosa y sus capas.
Figura 4. La imagen de la izquierda es un esquema de un cerebelo humano cortado por la línea media donde se señalan la sustancia blanca y la corteza cerebelosa (en rojo). La imagen de la derecha es un esquema de la corteza cerebelosa y sus capas.