La enfermedad de Huntington es una enfermedad hereditaria que lesiona células del cerebro. Las personas nacen con el gen defectuoso pero los síntomas no aparecen hasta después de los 30 o 40 años. Los síntomas iniciales de esta enfermedad pueden incluir movimientos descontrolados, torpeza y problemas de equilibrio , corea. Más adelante, puede impedir caminar, hablar y tragar. Algunas personas dejan de reconocer a sus familiares. Otros están conciente de lo que los rodea y pueden expresar sus emociones.
La enfermedad aparece en el 50% de la descendencia y es fácil de detectar tras un análisis de sangre en que se determine el gen de la enfermedad y si la desarrollará.
No existe una cura. Hay medicinas que pueden ayudarlo a controlar algunos síntomas, pero no pueden retrasar ni detener la enfermedad.
Es una enfermedad neurodegenerativa grave, pero relativamente rara, su prevalencia es de aproximadamente 2,7 personas por cada 100.000 1 . Es una enfermedad hereditaria con síntomas debilitantes, que generalmente se manifiesta en la edad adulta temprana y progresa con la edad. Todas las personas diagnosticadas con la enfermedad de Huntington han heredado al menos una copia del gen de la huntingtina mutante (HTT). La enfermedad afecta negativamente la función motora, psiquiátrica y cognitiva, hasta que el individuo no puede realizar tareas simples 2 .
Hasta la fecha, no existe tratamiento efectivos en de la enfermedad de Huntington
Los tratamientos actuales aprobados por la FDA para la enfermedad de Huntington simplemente actúan para aliviar un aspecto del perfil clínico y, se limitan a un tratamiento sintomático de la enfermedad. Los inhibidores del transporte vesicular de monoaminas (VMAT) , tetrabenazina y deutetrabenazina, son los ingredientes activos de los tratamientos aprobados por la FDA para el síntoma motor más común, la corea, una forma de discinesia caracterizada por movimientos espasmódicos involuntarios. Los estabilizadores del estado de ánimo, incluidos los antidepresivos, como la fluoxetina, y los antipsicóticos atípicos, como la risperidona , también se recetan comúnmente a los pacientes con enfermedad de Huntington.
Aunque el gen HTT se expresa de forma ubicua, el perfil clínico de la enfermedad de Huntington refleja la vulnerabilidad selectiva de HTT mutante (mHTT) en una vía cerebral conocida como circuito de los ganglios basales. Una clase de neuronas GABAérgicas estriatales, denominadas neuronas espinosas medianas (MSN), son un componente principal del circuito. Las MSN son una de las primeras células en sucumbir al estrés celular inducido por mHTT y sufrir apoptosis, seguidas de cerca por las neuronas corticales de proyección glutamatérgica 3 . La atrofia estriatal y cortical es una característica importante de la enfermedad de Huntington, junto con el ARN mHTT y los agregados de proteínas, que forman focos de ARN y cuerpos de inclusión, respectivamente, en la célula 2 .
Esta enfermedad hereditaria y genética tiene el gen mHTT diferente del alelo sano en que hay una expansión repetida inestable de una secuencia de tres nucleótidos, CAG, dentro del exón 1 de la forma mutante; esta secuencia de nucleótidos se conoce como microsatélite. Si la expansión de la repetición de microsatélites supera el umbral tolerado de 35 repeticiones CAG, el gen ahora codifica una transcripción mutante y la proteína 2 . En la transcripción del gen mHTT, el ARN mensajero (ARNm) y la proteína resultantes pueden formar cada uno una estructura terciaria anormal debido a su tracto poli-CAG y poli-glutamina, respectivamente. El ARNm y la proteína aberrantes de HTT causan estrés celular al desregular la transcripción y la traducción , induciendo deterioro mitocondrial e interrumpiendo el transporte citoplasmático nuclear 4,5,6. Además, la pérdida de la función HTT normal agrava aún más la patología celular, como lo demuestran los individuos homocigotos para el alelo mutante que desarrollan una progresión de la enfermedad más grave 7 . En última instancia, con el tiempo, el estrés celular se vuelve demasiado grande y la célula sufre apoptosis .
El perfil clínico de los pacientes con enfermedad de Huntington es muy variable, incluso entre individuos con el mismo número de repeticiones de CAG. Esta heterogeneidad interindividual, independiente de mHTT, llevó a la búsqueda de genes de riesgo más allá de HTT 8 . A través de grandes estudios de asociación de genoma de cohorte (GWAS), se identificaron polimorfismos de un solo nucleótido (SNP) específicos en los genes de reparación del ADN como influyentes importantes tanto en la edad de inicio como en la gravedad / velocidad de progresión de la enfermedad 8.9 . La solidez de este descubrimiento ha sido respaldada además por la investigación en modelos de roedores y células madre pluripotentes inducidas derivadas de pacientes (iPSC) 10,11,12 .
La inestabilidad somática es un fenómeno en el que la región expandida de las repeticiones CAG en el gen mHTT se expande aún más con el tiempo, como resultado de una reparación defectuosa del ADN. Durante la replicación y la transcripción del ADN, las regiones de secuencias repetitivas de ADN, en este caso repeticiones CAG, pueden plegarse sobre sí mismas para formar estructuras de horquilla / bucle de ADN cortas llamadas «ADN deslizado». Las proteínas de reparación de errores de emparejamiento reconocen la secuencia de ADN deslizada con errores de emparejamiento, pero durante la reacción de reparación, los bucles se incorporan a la secuencia del gen. Cada vez que se produce esta reparación de desajustes erróneos en las células, la región de repetición CAG crece más [ 13] . Como el proceso ocurre en las células somáticas, se acuñó el término inestabilidad somática. La actividad de proteínas reparadoras de ADN específicas.se sabe que influyen en la tasa de inestabilidad somática. Cuanto mayor sea la tasa de inestabilidad somática, más rápida será la progresión de la enfermedad en los sistemas modelo 11 . Además, las tasas de inestabilidad somática dependen del tipo de célula, y las neuronas MSN muestran una de las tasas más altas de inestabilidad somática. Esto explica en parte la vulnerabilidad selectiva de MSN en la enfermedad de Huntington.
Disminuir o detener la inestabilidad somática podría mitigar significativamente la progresión de la enfermedad y proporcionar una terapia modificadora de la enfermedad muy buscada. Por lo tanto, las proteínas de reparación del daño del ADN son de interés como nuevos objetivos farmacológicos en la enfermedad de Huntington 14 . Es de destacar la acción protectora de la nucleasa FAN1 frente a las reparaciones erróneas generadas por el complejo MSH3 / MSH2, MutSβ, reportado recientemente por Goold et al. 11 . Se están realizando investigaciones dentro de la industria farmacéutica para desarrollar moléculas pequeñas para modular estas proteínas reparadoras de desajustes, pero en el momento de redactar este artículo no hay ninguna disponible. Además, histonas desacetilasas(HDAC) se ha informado que reducen la inestabilidad somática en modelos de investigación, con el primer inhibidor de HDAC6 desarrollado en este contexto, CKD 504 de la compañía farmacéutica surcoreana Chong Kun Dang, que alcanzó los ensayos clínicos de fase I en 2018. Además, los informes iniciales de en Los modelos in vitro de la enfermedad de Huntington respaldarían una mayor investigación sobre el uso de inhibidores de la topoisomerasa 1 y de la tirosil-ADN fosfodiesterasa para ralentizar las tasas de expansión de la repetición CAG. Los inhibidores de la topoisomerasa ya han llegado a la clínica en terapias combinadas en oncología, pero su eficacia y tolerancia en la enfermedad de Huntington requieren más investigación 12 .
Las terapias usadas para la enfermedad de Huntington y otros trastornos de repetición de CAG
Hay alrededor de 50 enfermedades hereditarias conocidas causadas por expansiones de repetición de trinucleótidos y la enfermedad de Huntington es uno de al menos nueve trastornos de repetición CAG 15 . Aunque estas enfermedades van desde las que afectan la integridad muscular (distrofia miotónica), el control motor (esclerosis lateral amiotrófica) y la cognición (síndrome del X frágil), la inestabilidad somática es una característica compartida por todos. Las herramientas de investigación en esta área podrían tener un impacto significativo en el campo y acercarnos a una terapia común de modificación de la enfermedad.
ANTICUERPOS CONTRA EL CÁNCER La inmunoterapia para el cáncer es un resultado directo de los avances en la tecnología para la generación de anticuerpos recombinantes , la ingeniería de la terapia celular y genética y la creciente apreciación del papel del sistema inmunológico en la prevención y el control del cáncer. Se ha implementado con éxito en la clínica para cánceres hematológicos, como la leucemia linfocítica crónica (CLL), el linfoma no Hodgkin (NHL)y leucemia linfocítica aguda (ALL) . Si bien los tumores sólidos han demostrado ser más difíciles de atacar, la inmunoterapia está ganando terreno para el tratamiento en algunos casos. Una vez considerada un tratamiento de último recurso, la inmunoterapia suele ser la primera línea de tratamiento para el melanoma avanzado y metastásico . El sistema inmunológico participa en todas las etapas de la supresión tumoral: Las células asesinas naturales (NK) , las células T γδ y las células T CD8 citotóxicas promueven la apoptosis de las células tumorales. Las células dendríticas (DCs) prime CD4 y CD8 células T contra antígenos asociados a tumores (TAA). Los macrófagos fagocitan las células muertas y moribundas y promueven la inflamación . Sin embargo, durante la tumorigénesis y la progresión de la enfermedad, el tumor suprime activamente esta potente respuesta inmunitaria al: Creando un microambiente hipóxico, excluyendo las células T citotóxicas. Secreción de TGF-beta (TGF-β) y adenosina . Promover la formación y el reclutamiento de células inmunosupresoras: células T reguladoras (Tregs) , células supresoras derivadas de mieloides (MDSC) y macrófagos asociados a tumores (TAM). Regulación ascendente de ligandos inhibidores para prevenir la citotoxicidad mediada por células. En resumen, el sistema inmunológico tiene una potente respuesta antitumoral y el tumor tiene una potente respuesta antiinmunitaria. La inmunoterapia es lo que ayuda a inclinar la balanza a favor de la respuesta inmunitaria antitumoral. Inmunoterapia mediada por anticuerpos Una de las principales estrategias para estimular la respuesta inmune a los tumores aprovecha la especificidad de los anticuerpos. Según el Instituto de Investigación del Cáncer, hay tres tipos principales de terapias mediadas por anticuerpos: anticuerpos monoclonales (mAb), anticuerpos biespecíficos y conjugados de anticuerpo-fármaco (ADC). Anticuerpos monoclonicos Anticuerpos biespecíficos Descripción Reconoce un antígeno Reconoce dos antígenos Objetivo (antígeno) Marcadores de activación Marcadores inhibidores de las células inmunes Ligandos inhibidores en células tumorales Antígenos asociados a tumores (TAA) Receptor activador en células inmunes y TAA Dos puestos de control inmunes Función Agotamiento de células de leucemia y linfoma (ex Rituximab) Iniciar citotoxicidad mediada por células dependiente de anticuerpos (ADCC) Bloqueo de puntos de control inmunológico Redirección citotóxica: acerque las células inmunitarias y las células diana para facilitar la opsonización o la muerte celular. Co-bloqueo de puntos de control inmunológico: bloquea simultáneamente dos objetivos de puntos de control inmunitarios Nota: Puede encontrar más información sobre los ADC en el Instituto de Investigación del Cáncer.Volver a la cima Bloqueo de puntos de control inmunológico La respuesta inmune está estrictamente regulada por puntos de control, logrando un cuidadoso equilibrio entre los receptores inhibidores y de activación. Los receptores inhibidores de las células inmunitarias son fundamentales para evitar daños innecesarios al huésped. Las células innatas , como las células NK y las células T γδ, expresan de forma constitutiva receptores inhibidores para evitar la muerte y fagocitosis de las células hospedadoras sanas, mientras que las células T α / β regulan positivamente los receptores inhibidores poco después de la activación para regular la expansión y la función efectora. Las células tumorales, como las del cáncer de pulmón de células no pequeñas (NSCLC), el cáncer de próstata y el melanoma, regularán positivamente los ligandos inhibidores para evitar la destrucción por parte del sistema inmunológico. En general, la terapia de bloqueo de puntos de control inmunológico busca interrumpir la interacción receptor-ligando entre el receptor inhibidor de las células inmunes y el ligando expresado en las células tumorales. La terapia de bloqueo de puntos de control libera a las células inmunitarias de esta inhibición. La caracterización ex vivo o in vitro de los puntos de control inmunitarios es el primer paso hacia el desarrollo de una inmunoterapia eficaz. Además de la citometría de flujo , la inmunohistoquímica (IHC) y la inmunocitoquímica / inmunofluorescencia (ICC / IF) , Bio-Techne tiene una gran selección de anticuerpos de puntos de control inmunitarios altamente validados para el bloqueo y la neutralización. Datos de ELISA funcional que muestran un bloqueo exitoso de la interacción receptor-ligando con el anticuerpo bloqueador del receptor. La línea naranja muestra que el ligando recombinante se une al receptor de manera dependiente de la dosis, en ausencia del anticuerpo. (Izquierda) A 0.09-0.72 µg / mL, Mouse Anti-Human PD-1 (1015846) (Catálogo # MAB10864 ) bloqueará el 50% de la unión de 5 µg / mL de recombinante humano PD-L1 / B7-H1 Fc Chimera (línea naranja, catálogo nº 156-B7 ) a la proteína recombinante humana PD1 etiquetada con His inmovilizada (nº de catálogo 8986-PD ) recubierta a 1 µg / ml (100 µl / pocillo). A 5 µg / mL, este anticuerpo bloqueará> 90% de la unión. (Medio) A 70-350 ng / mL, conejo Anti-Cynomolgus TIGIT(2629A) (catálogo n.o MAB10532 ) bloqueará el 50% de la unión de TIGIT de mono Cynomolgus recombinante ( n.o de catálogo 9380-TG ) unido a CD155 / PVR humano recombinante inmovilizado ( n.o de catálogo 2530-CD ) recubierto a 2,5 µg / ml (100 µL / pocillo). (derecha) A 0.08-0.8 µg / mL, Rat Anti-Mouse CD47 (974222) (Catálogo # MAB18661 ) bloqueará el 50% de la unión de 0.25 µg / mL de Recombinant Mouse CD47 Fc Chimera (línea naranja, Catálogo # 1866- CD ) al ratón recombinante inmovilizado SIRP alpha / CD172a Fc Chimera ( n.o de catálogo 7154-SA) recubierto a 1 µg / mL (100 µL / pocillo). A 5 µg / mL, este anticuerpo bloqueará> 90% de la unión. Objetivos actuales y emergentes para el bloqueo de puntos de control inmunológico Las dos primeras terapias de bloqueo de puntos de control inmunitarios aprobadas para su uso en humanos se dirigen a los ejes CTLA-4 / CD80 – CD86 y PD-1 / PD-L1 , ambos miembros de las familias de puntos de control inmunitarios B7-CD28. Las proteínas de la familia B7 se expresan en la superficie de las células presentadoras de antígenos (APC) y las células tumorales e interactúan con los receptores de la familia de proteínas CD28 en las células inmunitarias. Los miembros de estas familias son necesarios para las funciones coestimuladoras y co-inhibidoras de las células T. Se están investigando muchos otros miembros de las familias de proteínas B7-CD28 como dianas adicionales para la inmunoterapia. PD-L1 es un objetivo clínico para el bloqueo de puntos de control inmunológico. Anticuerpo anti-PD-L1 altamente validado con reactividad cruzada en tejidos humanos, de ratón y de rata. ICC / IF de la línea celular U-251 de glioblastoma maligno. Las células se fijaron en paraformaldehído al 4% durante 10 minutos y se permeabilizaron en Triton X-100 al 0,05% en PBS durante 5 minutos. Las células se incubaron con Rabbit Anti-Human / Mouse / Rat PD-L1 (Catálogo # NBP1-76769 ) a 1 ug / ml durante la noche a 4C y se detectaron con un anti-conejo DyLight 488 (Green) a una dilución 1: 1000 para 60 minutos. Los núcleos se contratiñeron con DAPI (azul). Se tomaron imágenes de las células usando un objetivo de 100X y se desconvolucionaron digitalmente. NBP1-76769 ha sido validado por tres de los cinco pilares de la validación de anticuerpos , que incluyenEstrategias genéticas, estrategias de anticuerpos independientes y estrategias biológicas . B7 Puntos de control de inmunidad familiar Receptor: en células inmunes Ligando: en la célula tumoral PD-1PD-L1 , PD-L2CTLA-4CD80 , CD86 Receptor desconocido B7-H3 / CD276 Receptor desconocido B7-H4PSGL-1 , receptor alternativo desconocido VISTA / B7-H5VISTA / B7-H5VSIG-3KIR3DL3B7-H7 / HHLA2 Bloqueo / neutralización de HHLA-2 en un ensayo de citometría de flujo funcional. Brevemente, la línea celular de riñón embrionario humano HEK293 se transfectó con B7-H7 / HHLA-2 humano y se tiñó con TMIGD2 / CD28H humano recombinante biotinilado (10 ng / ml, catálogo n. ° 8316-TR ), seguido de estreptavidina-APC (catálogo n. ° F0050 -NOV ) (negro, línea de puntos). La unión está completamente bloqueada (histograma naranja) por 2,5 μg / mL de Mouse Anti-Human B7-H7 / HHLA2 (907812) (Catálogo # MAB80841 ). Se usó como control el control de isotipo IgG1 de ratón (nº de catálogo MAB002 ) a 2,5 μg / ml (línea azul). Un estudio de 2018 encontró que solo alrededor del 43% de los pacientes con cáncer son elegibles para la terapia de bloqueo de puntos de control, y la frecuencia de los pacientes que responden es de alrededor del 12%. Debido a que un porcentaje tan pequeño de pacientes responde al bloqueo de puntos de control actual, los puntos de control adicionales, más allá de la familia B7, son de gran interés, incluidos otros inhibidores de la citotoxicidad, como TIM-3 y TIGIT . Las células tumorales también pueden regular al alza los ligandos para inhibir la fagocitosis patrullando los macrófagos y las células mieloides. Estos ligandos, como CD24 y CD47 , se expresan en todas las células de mamíferos y son una señal de «no me comas» para el sistema inmunológico innato. Validación de Estrategias Ortogonales. TIM-3 es un receptor inhibidor expresado en la superficie de las células T y las células NK. El ARNm de TIM-3 se detectó en secciones de tejido de amígdala humana incluidas en parafina fijadas con formalina sondas con ACD RNAScope® Probe HAVCR2 (ACD Catalog # 560681) y teñidas con ACD RNAScope® 2.5 HD Detection Reagents-Red (imagen derecha, ACD Catalog # 32260). La sección de tejido adyacente se procesó para inmunohistoquímica utilizando Anticuerpo TIM-3 antihumano de cabra (R&D Systems Catalog # AF2365 ) seguido de incubación con Anticuerpo de polímero VisUCyte HRP de IgG anti-cabra (R&D Systems, Catálogo # VC004 ) y cromógeno DAB (imagen derecha , amarillo marron). Los tejidos se tiñeron por contraste con hematoxilina (azul). Receptor: en células inmunes Ligando: en la célula tumoral Inhibición de la citotoxicidad LAG-3MHC-II , Galectin-3 , LSECtin / CLEC4G , FGL-1TIGITCD155 (PVR) , CD112 (PVRL2)TIM-3Galectina-9 , HMGB-1ILT2HLA-GNKG2AHLA-E Inhibición de la fagocitosis Siglec-10CD24SIRP-alfaCD47 Obtenga más información sobre objetivos de puntos de control inmunológicos con el libro electrónico de Bio-Techne, Objetivos de puntos de control inmunológicos actuales y emergentes para la investigación en inmuno-oncología . La expresión de HLA-G contribuye a la función inmunomoduladora de los queratinocitos humanos. Expresión de superficie de HLA-G determinada por citometría de flujo de queratinocitos humanos teñidos con Alexa Fluor 700 conjugado de ratón anti-HLA-G humano (87G) (número de catálogo NBP1-43123AF700 ) (histograma rojo) o control de isotipo (histograma azul). Imagen adaptada de Mestrallet G, Auvré F, Schenowitz C, Carosella ED, LeMaoult J, Martin MT, Rouas-Freiss N, Fortunel NO. Los queratinocitos humanos inhiben la proliferación de células T CD4 + a través de la secreción de TGFB1 y la expresión superficial de los puntos de control inmunológico HLA-G1 y PD-L1 . Células. 8 de junio de 2021; 10 (6): 1438. doi: 10.3390 / cells10061438. PMID: 34201301; PMCID: PMC8227977. Licencia proporcionada por CC-BY . La eficacia del bloqueo de los puntos de control inmunitarios está determinada por varios factores: la expresión de receptores inhibidores por parte de los tumores, la penetración de anticuerpos terapéuticos en los tumores sólidos y la accesibilidad del tumor a las células inmunitarias. Debido a la redundancia de la expresión del inhibidor (por ejemplo, tanto LAG-3 como PD-1 expresados en la misma célula) y la complejidad del microambiente tumoral (TME), podría ser necesario un bloqueo de puntos de control inmunitario combinado para lograr una inmunidad antitumoral óptima. El bloqueo combinado de puntos de control podría incluir dos o más receptores co-inhibidores o un enfoque de múltiples frentes dirigido a un receptor inhibidor y un inhibidor de la fagocitosis, como CD47 . El bloqueo combinado se puede lograr con un cóctel de anticuerpos monoclonales o un anticuerpo biespecífico diseñado. Receptor inhibidor de LAG-3 expresado en la superficie de células T estimuladas. Las células mononucleares de sangre periférica humana (PBMC) no se trataron (panel inferior) o se trataron con 5 μg / ml de PHA (panel superior). Las PBMC se tiñeron con Mouse Anti-Human LAG-3 (1009611) (Catálogo # MAB23195 ) seguido de Anticuerpo secundario anti-IgG de ratón conjugado con Alophycocyanin (APC) (Catálogo # F0101B ) y Anti-Human CD3 epsilon de ratón conjugado con PE (UCHT1) ) (Catálogo # FAB100P ). Los marcadores de cuadrante se establecieron basándose en la tinción del anticuerpo de control de isotipo (catálogo nº MAB003 ). HCDM utilizó el clon UCHT1 de CD3 en el taller de HLDA para establecer la designación de CD. Los avances en la ingeniería de anticuerpos han llevado al desarrollo de anticuerpos biespecíficos. Como lo indica su nombre, las regiones variables de estos anticuerpos reconocen dos antígenos diferentes. Estructura Ig-como F (ab) 2 ScFv Cadena pesada completa (Hc) y cadena ligera (Lc) de dos anticuerpos monoclonales Fragmentos de unión a antígeno de longitud completa (F (ab)) de dos anticuerpos monoclonales, ligados químicamente Fragmentos variables de cadena única (scFv) de la cadena pesada y ligera de dos anticuerpos monoclonales Función Se une a dos antígenos y la región constante (Fc) puede activar receptores Fc (FcR) en DC, macrófagos y células NK Bifuncional: une dos antígenos sin dominio Fc funcional Bifuncional: une dos antígenos sin dominio Fc funcional Ventajas Puede activar FcR para una funcionalidad adicional: ADCC Fagocitosis Talla pequeña Baja inmunogenicidad Agregar funcionalidad en las regiones del vinculador Talla pequeña Baja inmunogenicidad Puede diseñarse fácilmente para ser tri-específico Agregar funcionalidad en las regiones del vinculador El azul y el verde representan dos especificidades antigénicas diferentes. Sólo se muestra un ejemplo de estructuras de fragmentos F (ab) 2 y ScFv. Para la terapia del cáncer, los anticuerpos biespecíficos se utilizan de dos formas: Bloqueo simultáneo de dos controles inmunes. Redirección de células citotóxicas para interactuar con la célula tumoral. Enfoque: redirección de células citotóxicas Los anticuerpos biespecíficos para redirigir las células citotóxicas son de gran interés para la inmunoterapia del cáncer. El objetivo principal de este tipo de inmunoterapia es acercar la célula citotóxica, ya sea célula T o célula NK, a la célula tumoral para facilitar su destrucción. Idealmente, el anticuerpo biespecífico está diseñado para activar la célula citotóxica a través de CD3 para las células T y CD16 para las células NK y unirse a un TAA expresado de forma única. Antígeno asociado a tumor (TAA) Tipo de cáncer Antígeno carcinoembrionario (CEA) Cáncer colonrectal CD33 / Siglec-3 Leucemia mieloide aguda (AML), linfomas EGFR Cáncer de mama, cáncer de páncreas HER2 / ERBB2 Cáncer de mama, cáncer de próstata EpCAM Cáncer de mama, cáncer colorrectal, cáncer de endometrio, cáncer de ovario, cáncer de páncreas, cáncer de próstata MUC1 Cáncer de mama, cáncer de ovario, cáncer de próstata Receptor de prolactina (PRLR) Cáncer de mama VEGFR1 Cáncer de mama La expresión de VEGFR1 / Flt-1 se limita a las células cancerosas y no se expresa en las células del estroma tumoral. Análisis IHC-Parafina (IHC-P) de una sección de tejido de carcinoma de mama humano embebido en parafina (FFPE) fijada con formalina usando una dilución 1: 500 de Anticuerpo Policlonal Anti-VEGFR1 / Flt-1 de Conejo (Catálogo # NB100-527 ). El ensayo implicó 20 minutos de recuperación de antígeno inducida por calor (HIER) con tampón de citrato de sodio 10 mM (pH 6,0) y extinción de peroxidasa endógena usando bloque de peróxido. Se utilizó un sistema de detección refinado con polímero y DAB para la detección de la señal que siguió a la contratinción con hematoxilina. La tinción se realizó con Histowiz . A menudo, los TAA dirigidos por la terapia con anticuerpos no son exclusivos de las células tumorales y también se expresan en células sanas. Sin embargo, los TAA se sobreexpresan con frecuencia en las células tumorales en comparación con las células sanas, lo que brinda la oportunidad de aprovechar la tecnología avanzada de ingeniería de anticuerpos. Para limitar la redirección de células citotóxicas a células sanas no malignas, los anticuerpos biespecíficos proporcionan una clara ventaja sobre las terapias de anticuerpos tradicionales debido a una técnica de ingeniería a la que a menudo se hace referencia como ajuste de afinidad. El ajuste de afinidad implica la selección de anticuerpos con ciertas propiedades de unión, o afinidad, por un antígeno particular. Durante una respuesta inmune in vivo, los anticuerpos a menudo se someten a un proceso llamado maduración por afinidad en el que se seleccionan anticuerpos de alta afinidad en la reacción del centro germinal. Un enfoque de ajuste de afinidad permite la selección de anticuerpos biespecíficos con unión de anticuerpo débil (baja afinidad) al receptor de activación (CD3 o CD16) y unión fuerte (alta afinidad) a TAA, como EpCAM. Esto aumenta la probabilidad de que el anticuerpo se una a una célula tumoral antes de que active la célula citotóxica, mejorando la eficacia y reduciendo la activación no específica. Los anticuerpos Bio-Techne están validados para una variedad de aplicaciones, incluido ICC / IF. Análisis inmunofluorescente confocal de la línea celular de cáncer de ovario SK-OV-3 usando EpCAM antihumano de ratón conjugado con Alexa Fluor 488 (EGP40 / 826) (catálogo nº NBP2-44643 ) (verde). Los filamentos de actina F se marcaron con DyLight 554 Phalloidin (rojo). Se usó DAPI para teñir los núcleos celulares (azul). Como desafío adicional, los anticuerpos dirigidos contra CD3 se dirigirán indiscriminadamente tanto a las células T citotóxicas CD8 + como a las células reguladoras T CD4 +, lo que podría promover una TME inmunosupresora. Aunque las terapias actualmente aprobadas se dirigen a CD3 y TAA, los investigadores también están explorando anticuerpos biespecíficos dirigidos a marcadores de células NK, como CD16A . CD16 se expresa en la superficie de las células NK humanas. Se tiñeron células mononucleares de sangre periférica humana (PBMC) con Fc gamma RIII / CD16 antihumano de ratón conjugado con PE (245536) (nº de catálogo FAB2546P ) y CD14 antihumano de ratón conjugado con APC (134620) (nº de catálogo FAB3832A ). Los marcadores de cuadrante se establecieron basándose en la tinción del anticuerpo de control de isotipo (nº de catálogo IC003P ). Limitaciones y direcciones futuras de la inmunoterapia basada en anticuerpos Con la implementación de terapias basadas en anticuerpos, ha habido avances significativos en el tratamiento del cáncer de neoplasias malignas de alta mortalidad, como NSCLC metastásico y melanoma. Sin embargo, todavía existen limitaciones significativas de la inmunoterapia mediada por anticuerpos, que incluyen: Penetración limitada en tumores sólidos. Los anticuerpos de tipo Ig con dominios Fc pueden ser inmunogénicos. El bloqueo de puntos de control solo es eficaz para tumores «calientes» con actividad inmunitaria y expresión de ligandos inhibidores. Nanoanticuerpos ® con anticuerpos VHH de dominio único, como de Bio-Techne LlaMABodies TM , pueden dirigirse a epítopos de difícil acceso, y pueden tener un papel en inmunoterapias futuras. REFERENCIAS Moore GL, Lee SH, Schubbert S, Miranda Y, Rashid R, Pong E, et al. El ajuste de la afinidad de las células T mejora la eficacia y la seguridad de los anticuerpos biespecíficos anti-CD38 × anti-CD3 en monos, una terapia potencial para el mieloma múltiple. Blood 2015; 126 : 1798-1798. https://doi.org/10.1182/BLOOD.V126.23.1798.1798 . Wang W, Guo H, Geng J, Zheng X, Wei H, Sun R, et al. La galectina-3 liberada por el tumor, un ligando inhibidor soluble de la NKp30 humana, desempeña un papel importante en el escape del tumor del ataque de las células NK. J Biol Chem 2014; 289 : 33311. https://doi.org/10.1074/JBC.M114.603464 . Lum LG, Thakur A, Choi M, Deol A, Kondadasula V, Schalk D, et al. Respuestas clínicas e inmunes a células T activadas armadas con anticuerpos biespecíficos anti-CD3 x anti-EGFR (EGFR BAT) en pacientes con cáncer de páncreas. Https: // DoiOrg / 101080 / 2162402X20201773201 2020; 9 :. https://doi.org/10.1080/2162402X.2020.1773201 . Müller P, Rios-Doria J, Harper J, Cao A. Combinación de ADC con agentes de inmuno-oncología. Desarrollo de descubrimiento de fármacos contra el cáncer 2018: 11–44. https://doi.org/10.1007/978-3-319-78154-9_2 . Chauvin JM, Zarour HM. TIGIT en inmunoterapia contra el cáncer. J Immunother Cancer 2020; 8 : e000957. https://doi.org/10.1136/JITC-2020-000957 . Barkal A, Brewer R, Markovic M, Kowarsky M, Barkal S, Zaro B, et al. La señalización de CD24 a través del macrófago Siglec-10 es un objetivo para la inmunoterapia contra el cáncer. Naturaleza 2019; 572 : 392–6. https://doi.org/10.1038/S41586-019-1456-0 . Strome AL, Zhang X, Strome SE. El papel evolutivo de la inmuno-oncología para el tratamiento del cáncer de cabeza y cuello. Laringoscopio Investig Otolaryngol 2019; 4 : 62–9. https://doi.org/10.1002/LIO2.235 . Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Células dendríticas en inmunología e inmunoterapia del cáncer. Nat Rev Immunol 2019 201 2019; 20 : 7-24. https://doi.org/10.1038/s41577-019-0210-z . Veglia F, Sanseviero E, Gabrilovich DI. Células supresoras derivadas de mieloides en la era de la creciente diversidad de células mieloides. Nat Rev Immunol 2021 218 2021; 21 : 485–98. https://doi.org/10.1038/s41577-020-00490-y . DeNardo DG, Ruffell B. Los macrófagos como reguladores de la inmunidad e inmunoterapia tumoral. Nat Rev Immunol 2019 196 2019; 19 : 369–82. https://doi.org/10.1038/s41577-019-0127-6 . Labrijn AF, Janmaat ML, Reichert JM, Parren PWHI. Anticuerpos biespecíficos: una revisión mecanicista de la tubería. Nat Rev Drug Discov 2019188 2019; 18 : 585–608. https://doi.org/10.1038/s41573-019-0028-1 . Waldman AD, Fritz JM, Lenardo MJ. Una guía para la inmunoterapia del cáncer: de la ciencia básica de las células T a la práctica clínica. Nat Rev Immunol 2020 2011 2020; 20 : 651–68. https://doi.org/10.1038/s41577-020-0306-5 . Marshall HT, Djamgoz MBA. Inmuno-oncología: objetivos emergentes y terapias combinadas. Front Oncol 2018; 0 : 315. https://doi.org/10.3389/FONC.2018.00315 . Nanobodies es una marca registrada de Ablynx NV
Don Santiago descubrió una hendidura en el retículo que formaba el sistema nervioso. Con ello rompió la teoría sincitial del sistema nervioso y creo la teoría de la neurona.
Tambien el sistema nervioso esta compuestos de células. “Llamadas “Neuronas” A la hendidura que descubrió Don Santiago , Sherrington las llamo Sinapsis ,
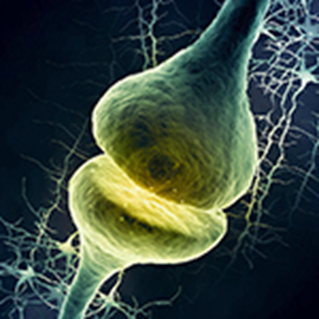
Una sinapsis es una estructura que permite a una neurona pasar una señal eléctrica o química a otra célula y asi transmitir los impulsos nerviosos de una neurona a otra y constituye la base del funcionamiento del sistema nervioso.
En una sinapsis, la neurona presináptica está muy próxima a la membrana de otra celula , la postsináptica y las separada un espacio conocido como hendidura sináptica.
La complejidad de este espacio sostienen una compleja maquinaria molecular que unen las dos membranas y llevan a cabo el proceso de señalización. En la porción presináptica destacan las proteínas implicadas en el almacenamiento, liberación y recaptación del neurotransmisor y en la postsináptica, los receptores, que son proteínas específicas a las que se une el transmisor.
En muchas sinapsis, la parte presináptica se encuentra en un axón y la parte postsináptica en una dendrita o soma. Muchas dendritas tienen expansiones de su superficie, con aspecto de chupa-chups que son característicos lugares postsinápticos y se conocen como espinas dendríticas.
Las espinas dendríticas se pueden contar con un microscopío óptico, y tener una estimación realista de los cambios en el número de sinapsis. Dado que aproximadamente el 90 por ciento de las sinapsis excitatorias se localizan en las espinas dendríticas, los cambios en su número y morfología podrían afectar a la regulación de la transmisión sináptica y a la plasticidad estructural neuronal en general y pueden estar relacionados con la comunicación cerebral atípica que muestran las personas con trastornos del espectro del autismo (TEA).
Durante el desarrollo temprano, el cerebro produce un exceso de sinapsis y posteriormente elimina aquellas que no son necesarias, no están bien o están inactivas en un proceso que se conoce como «pruning» o poda sináptica. Esas sinapsis son eliminadas en un proceso de autofagia, en el que unos elementos cerebrales comen las sinapsis alteradas o inactivas. El desarrollo postnatal de la sinapsis en la corteza cerebral de los mamíferos es un proceso dinámico que implica que la formación y la poda son simultáneas. A edades tempranas, la formación de sinapsis supera a la poda, lo que da lugar a un exceso de sinapsis excitatorias, esenciales para la estructuración de los circuitos neuronales. Posteriormente, la eliminación de sinapsis supera a la formación, dando lugar a una poda neta de espinas desde la infancia hasta la adolescencia. La densidad de espinas dendríticas alcanza su punto máximo en la primera infancia y va seguida de un pronunciado descenso durante la última etapa de la infancia y la adolescencia hasta alcanzar los niveles de los adultos. Este proceso causa la selección y la maduración de las sinapsis y los circuitos neuronales.
Los procesos de diferenciación de las células neurales y la formación de circuitos mediante contactos sinápticos entre neuronas (sinaptogénesis) ocurren en el sistema nervioso central durante las últimas fases del desarrollo prenatal y los primeros meses después del nacimiento [1,2]. Ambos procesos requieren la participación de múltiples mecanismos moleculares y celulares organizados en patrones espaciotemporales específicos, cuya alteración es la base para la aparición de anomalías funcionales, con el resultado de enfermedades psiquiátricas asociadas al neurodesarrollo. La consecuencia de alteraciones en estos procesos es una anomalía en la función de los circuitos neuronales, es decir, en el patrón de conexiones de las neuronas entre algunas regiones del cerebro o de la funcionalidad de las sinapsis entre las neuronas que conforman estos circuitos. Estas alteraciones tendrán como consecuencia un desequilibrio entre la actividad excitatoria (incremento de actividad) e inhibitoria (disminución de actividad) de las sinapsis en los circuitos afectados.
Desde que el niño nace y hasta la adolescencia su cerebro está en continua evolución. En esa etapa se generan muchas sinapsis neuronales como resultado de los aprendizajes que va adquiriendo, a la vez que se entrena la plasticidad neuronal, que no es más que la capacidad del cerebro para reorganizarse y formar nuevas conexiones neurales con el objetivo de adaptarse al entorno.
En estos años también tiene lugar un proceso muy importante en el cerebro infantil que permite reorganizar la estructura sináptica y optimizar el procesamiento de la información: la poda sináptica o poda regulatoria.
Durante los primeros años de vida, en el cerebro infantil se crean nuevas sinapsis a una velocidad sorprendente que puede alcanzar hasta las 40.000 conexiones neurales por segundo. Esto le permite al niño conocer su entorno y adquirir nuevos conocimientos a una gran velocidad, el problema es que con el paso del tiempo muchos de esos aprendizajes dejan de ser útiles. De esta manera, en el cerebro infantil se mantienen muchas sinapsis que no son funcionalmente necesarias y que en lugar de facilitar el procesamiento de la información, lo ralentizan.
Para evitar que esto ocurra y garantizar un procesamiento eficiente de la información tiene lugar la poda sináptica, que no es más que el proceso mediante el cual se eliminan las conexiones más débiles entre las neuronas que se crearon durante los primeros años de vida. Básicamente, se trata de un proceso regulador que garantiza una organización sináptica más eficiente, eliminando las estructuras sinápticas innecesarias del cerebro para aumentar la superficie de recepción de los neurotransmisores.
Por lo general, el período de poda comienza alrededor del segundo año de vida y se extiende hasta avanzada la adolescencia. La poda en las zonas de la corteza involucradas con la percepción visual y auditiva suele completarse alrededor del 4 y 6 año de vida mientras que la poda de las funciones superiores como el control inhibitorio y la autorregulación emocional continúa hasta los primeros años de la juventud.
Hasta ese momento, la poda regulatoria puede haber reducido entre un 30 o 40% el número de sinapsis en el cerebro, lo que significa que el niño habrá perdido algunas de las habilidades o conocimientos que aprendió en algún momento de la infancia y que dejó de utilizar. En su lugar se habrán creado nuevas conexiones correspondientes a las nuevas habilidades que va desarrollando.
La poda sináptica es tan importante para el desarrollo cerebral como la creación de nuevas conexiones ya que de esta manera se eliminan del cerebro las sinapsis que no se utilizan, se refuerzan las estructuras neuronales más importantes y se da paso a la creación de nuevas conexiones. Sin embargo, más allá de su importancia anatómica, la poda sináptica también beneficia el desarrollo cognitivo, emocional y conductual de los niños.
La poda al eliminar conexiones inútiles, favorece la consolidación de una red cerebral mucho más eficiente que permite que se creen nuevas sinapsis con mayor facilidad, y Contribuye a la adaptación del entorno. Durante la poda cerebral desaparecen las sinapsis que el niño ya no utiliza para dejar paso a la creación de nuevas conexiones que le permitan adaptarse con más facilidad a su nuevo entorno, como reveló un estudio realizado en la Escuela de Medicina Harvard.
Los expertos consideran la poda sináptica como un proceso madurativo en el que se consolidan las conexiones cerebrales y las funciones cognitivas más importantes en los niños. En especial, la poda de la sustancia gris, sobre todo en las áreas de la corteza cerebral, se ha relacionado con un mayor desarrollo de funciones como la memoria, el control atencional y la concentración.
La poda sináptica se ha relacionado con la creciente y rápida adquisición de habilidades y conocimientos que tiene lugar hasta avanzada la adolescencia. En cambio, las alteraciones en este proceso se han vinculado con algunos trastornos del desarrollo como el autismo, según reveló una investigación realizada en la Universidad Icesi.
La poda sináptica está fuertemente influenciada por factores ambientales y del desarrollo infantil. Básicamente, aquellas funciones que el niño necesita para desenvolverse en su entorno serán las que se conserven mientras que las habilidades que ya no le sean útiles o haya dejado de ejercitar serán las que desaparezcan. Por eso, es importante poner en práctica algunas estrategias para estimular el proceso de poda sináptica y reforzar las habilidades que se quieren preservar.
Un grupo de investigadores italianos (Pagani et al., 2021) ha publicado un artículo en la revista Nature Communications en el que señalan que hay un exceso de sinapsis en la corteza cerebral de las personas con autismo.
Muchos de los genes implicados en el autismo intervienen en el control de la traducción y la síntesis de proteínas, pero también en la estructura, funcionamiento y plasticidad de las sinapsis. Múltiples genes de susceptibilidad al TEA convergen en vías celulares que están implicadas en el sitio postsináptico de las sinapsis glutamatérgicas.
Un estudio previo (Hutsler y Zhang, 2010) había encontrado una mayor densidad de espinas dendríticas en las células piramidades de la corteza cerebral de las personas con TEA en comparación con casos control de las mismas edades. Los dos investigadores estudiaron las células piramidales de las capas corticales superficiales y profundas de los lóbulos frontal, temporal y parietal. Las mayores densidades de espinas aparecían predominantemente en la capa II de las distintas regiones corticales y en la capa V del lóbulo temporal. El aumento de la densidad de espinas iba asociado a pesos cerebrales reducidos y se encontraron con mayor frecuencia en personas con TEA con niveles más bajos de funcionamiento cognitivo
La poda sináptica es tan importante para el desarrollo cerebral como la creación de nuevas conexiones ya que de esta manera se eliminan del cerebro las sinapsis que no se utilizan, se refuerzan las estructuras neuronales más importantes y se da paso a la creación de nuevas conexiones. Sin embargo, más allá de su importancia anatómica, la poda sináptica también beneficia el desarrollo cognitivo, emocional y conductual de los niños.
También se han visto cambios en la estructura sináptica y número de sinapsis en múltiples modelos de ratón para el TEA, incluidos los modelos de ratón del síndrome de Rett, del X frágil y de la duplicidad 15q11-13, así como los ratones deficientes en los genes CYFIP1 (proteína citoplasmática que interactúa con FMR1), CNTNAP2 (proteína similar a la contactina), DLGAP2 (proteína asociada a DLG 2) y SHANK1 (dominios SH3 y múltiples repeticiones de anquirina 1). Sin embargo, no siempre son resultados en el mismo sentido, que indiquen un aumento de espinas y de sinapsis en los ratones «con autismo» frente a los animales control. La cepa de ratones C58/J muestra una baja sociabilidad, deterioro de la comunicación y comportamiento estereotipado; por lo tanto, se considera entre los modelos animales adecuados para el estudio del autismo idiopático. Un estudio de investigadores mexicanos ha analizado las diferencias en el número y morfología de espinas dendríticas en el hipocampo y la corteza prefrontal de los ratones C58/J. Los investigadores encontraron cambios en el número de espinas y en la morfología de forma dependiente de la región cerebral: una sutil disminución de la densidad de espinas en el córtex prefrontal, una mayor frecuencia de espinas de fenotipo inmaduro caracterizadas por una longitud similar a la de los filopodios o una morfología pequeña, y un menor número de espinas de fenotipo maduro en el hipocampo. Estas diferencias entre lo encontrado en estos ratones y lo observado en tejido cerebral humano ejemplifican las dificultades de utilizar modelos murinos para un trastorno como los TEA.
Las observaciones llevadas a cabo hasta el momento en personas con TEA han llevado a la hipótesis de que una poda sináptica disfuncional y una homeostasis alterada podrían contribuir a la patología del TEA. Los exámenes histológicos postmortem apoyan esta idea, ya que se ha observado repetidamente la presencia de una mayor densidad de espinas dendríticas en el tejido cerebral de los pacientes con TEA.
Las mayores densidades de espinas en las personas con TEA proporcionan apoyo estructural a la hipótesis que plantea la existencia de cambios en los circuitos sinápticos de la corteza cerebral y que estas modificaciones pueden generar un funcionamiento alterado de la actividad cerebral.
ANATOMÍA CEREBRAL EN EL AUTISMO
Genéricamente lo conocemos todos , El crecimiento del manto cerebral , sus conexiones y sus núcleos de agrupación celular, son imprescindible, para la función cerebral, que aumenta y disminuyen con la evolución.
Sin embargo lo que dificulta el entendimiento, es como estas lesiones anatómicas o funcionales proporcionan estos daños.
Es el esquema de siempre, como esta materia produce este espiritu
En el desarrollo típico, del cerebro, la corteza cerebral, se engrosa hasta aproximadamente los 2 años y luego se vuelve gradualmente más delgada hasta la adolescencia a medida que el cerebro madura. El nuevo estudio, uno de los más grandes para investigar el grosor cortical en el autismo, se alinea con otros que indican que esta trayectoria difiere en las personas con la afección.
Los cambios mas objetivos, no dicen absolutamente nada, sobre todo si los comparamos con grades cambios y lesiones del cerebro en superdotados. Hay algo mas. Pero eso lo sabíamos de siempre
Los resultados sugieren que la estructura cerebral no cambia de manera uniforme en el autismo, sino que varía con factores como la edad, el sexo y el coeficiente intelectual, Canadá. Un estudio reciente encontró que varias regiones de la corteza cerebral son más gruesas en niños y adultos jóvenes con autismo que en sus pares de desarrollo típico.
Las diferencias son mayores en las niñas, en niños de 8 a 10 años y en aquellos con un bajo coeficiente intelectual (IQ)
Conocer los procesos neurales ligados a la formación de sinapsis y circuitos cerebrales para entender su papel en las enfermedades del neurodesarrollo, como el trastorno del espectro autista (TEA) y el trastorno por déficit de atención/hiperactividad (TDAH).
Desarrollo. La actividad de los circuitos neuronales es la base neurobiológica de la conducta y la actividad mental (emociones, memoria y pensamientos). Los procesos de diferenciación de las células neurales y la formación de circuitos por contactos sinápticos entre neuronas (sinaptogénesis) ocurren en el sistema nervioso central durante las últimas fases del desarrollo prenatal y los primeros meses después del nacimiento. Los TEA y el TDAH comparten rasgos biológicos, relacionados con alteraciones en los circuitos cerebrales y la función sináptica, que permiten tratarlos científicamente de forma conjunta. Desde el aspecto neurobiológico, el TEA y el TDAH son manifestaciones de anomalías en la formación de circuitos y contactos sinápticos en regiones cerebrales implicadas en la conducta social, especialmente en la corteza cerebral prefrontal. Estas anomalías son causadas por mutaciones en genes involucrados en la formación de sinapsis y plasticidad sináptica, la regulación de la morfología de las espinas dendríticas, la organización del citoesqueleto y el control del equilibrio excitador e inhibidor en la sinapsis.
Conclusiones. El TEA y el TDAH son alteraciones funcionales de la corteza cerebral, que presenta anomalías estructurales en la disposición de las neuronas, en el patrón de conexiones de las columnas corticales y en la estructura de las espinas dendríticas. Estas alteraciones afectan fundamentalmente a la corteza prefrontal y sus conexiones.
Por lo tanto, es importante conocer los procesos neurales ligados a la actividad de los circuitos cerebrales para entender las consecuencias de su disfunción y, con ello, su papel en el desarrollo de los síntomas característicos de las enfermedades del neurodesarrollo, como son los trastornos del espectro autista (TEA) y el trastorno por déficit de atención/hiperactividad (TDAH) [3].
El TEA es una condición heterogénea caracterizada por la presencia de alteraciones del comporamiento en la interacción social y comunicación, acompañada de comportamiento estereotipado e intereses restringidos. Además de estos síntomas necesarios para el diagnóstico, el TEA a menudo se presenta con una variedad de otras manifestaciones conductuales y funcionales, como problemas de lenguaje, hiperactividad, epilepsia, déficit de atención y trastornos del sueño. El TDAH se inicia en la infancia y se caracteriza por dificultades para mantener la atención, hiperactividad con exceso de movimiento e impulsividad, y dificultades en el control de los impulsos. El TEA y el TDAH comparten rasgos neurobiológicos, fundamentalmente relacionados con alteraciones en la estructura y función de la corteza cerebral, que permiten tratarlos conjuntamente.
La actividad de los circuitos neuronales es la base neurobiológica de los procesos del sistema nervioso central que se manifiestan en la conducta y los procesos mentales (emociones, memoria y pensamiento). La función de los circuitos presenta una importante capacidad de adaptación, mediante cambios en las propiedades espaciales y temporales de las conexiones entre neuronas del circuito. Así, el cerebro construye una respuesta adecuada a los requerimientos de cada situación interna o ambiental. La base estructural de la adaptación neural es la capacidad de modificar la cantidad y la función de las sinapsis neuronales; por lo tanto, lo que definimos como plasticidad neural (neuroplasticidad) se fundamenta en la plasticidad sináptica en los circuitos neuronales [2,4]. La maleabilidad funcional se logra durante el desarrollo modulando la expresión de un conjunto de genes que regulan mecanismos moleculares y celulares que influyen en la dinámica de las conexiones sinápticas. La neuroplasticidad durante el desarrollo del cerebro presenta patrones temporales heterogéneos: existe un período crítico de mayor maleabilidad sináptica alrededor del nacimiento, que modula la regulación génica para la formación y consolidación de conexiones neuronales adecuadas mediante la influencia de los estímulos ambientales. Éstos actúan sobre un patrón de conexiones regulado por la información genética (lo que hace que los humanos generemos un cerebro humano).
TEA y el TDAH pueden ser manifestaciones de anomalías en el proceso de neuroplasticidad del desarrollo, al igual que otros trastornos neuropediátricos congénitos y adquiridos, como la encefalopatía por hipoxia neonatal, parálisis cerebral, epilepsia, distonía, discapacidad intelectual y esquizofrenia [5,6]. Desde su aspecto etiológico, ambos procesos tienen una importante carga genética, considerándose trastornos poligénicos (múltiples genes implicados con carga patogénica escasa y variable) y, por tanto, derivados de una combinación de alteraciones genéticas de novo (mutaciones espontáneas) asociadas a una predisposición derivada de variaciones comunes heredadas. Las principales anomalías genéticas asociadas a TEA y TDAH implican genes que codifican proteínas de la sinapsis [3,7].
Los conocimientos acumulados en los últimos años muestran que las enfermedades mentales de inicio en la infancia se deben a alteraciones de la formación o de la actividad de circuitos neuronales. Entre estas enfermedades destacan el TEA y el TDAH, asociados o no a discapacidad intelectual, y otros síndromes del neurodesarrollo. La aparición durante la vida temprana de las alteraciones conductuales y funcionales del TEA y el TDAH induce Es lógico pensar que las alteraciones anatómica y fisiológica están producido por una previa alteración cromosómica. Nada en nuestra biología aparece o desaparece sin la alteración genética .
En el TEA y al TDAH se modifican los procesos del desarrollo neuronal y el establecimiento de conexiones, sobrepasando la capacidad compensatoria de la neuroplasticidad del sistema nervioso central durante el desarrollo y generando alteraciones en el patrón inicial de conexiones en los circuitos neuronales.
Durante el desarrollo embrionario, los axones de las neuronas jóvenes llegan a su destino mediante procesos bien regulados de guía axonal, estableciendo conexiones inmaduras y temporales con las neuronas que están diferenciándose en las regiones diana. Durante el desarrollo de las conexiones en la corteza cerebral aparece una estructura transitoria, la subplaca, que se forma entre los 3-4 meses de desarrollo y constituye el principal compartimiento de conexión neuronal de la corteza hasta los siete meses. La subplaca desaparece progresivamente, en la etapa posnatal temprana, hasta los seis meses de vida [8]. Las fibras nerviosas que van a establecer contactos en la corteza entran primero en la subplaca y establecen circuitos sinápticos temporales, donde permanecen un ‘tiempo de espera’ antes de entrar en la placa cortical para establecer sinapsis con las neuronas de las diferentes capas corticales. Desde los siete meses de desarrollo hasta un año de vida posnatal, la subplaca es un lugar de relevo sináptico. Estas sinapsis transitorias desarrollan circuitos neuronales transitorios, que representan la base neurobiológica de la actividad eléctrica del comportamiento fetal y de los neonatos prematuros. Durante la etapa perinatal se extienden las fibras corticales desde la subplaca hacia las neuronas de la placa cortical (futura corteza cerebral), con lo que se inicia y progresa la formación de circuitos de conexión maduros entre áreas de la corteza cerebral. Se origina entonces una sobreproducción sináptica que permanece en la infancia, en la que los procesos de generación predominan sobre los de retracción sináptica, hasta llegar a la adolescencia, donde se invierte el patrón y se produce una poda selectiva de los contactos no funcionales (es decir, predomina la eliminación de sinapsis poco eficaces sobre la generación de nuevas) (Figura). El equilibrio entre producción y eliminación sináptica seguirá extendiéndose a lo largo de la vida y es lo que denominamos neuroplasticidad adaptativa y reactiva. Recientemente se ha podido demostrar que las neuronas de la subplaca se relacionan embriológica y funcionalmente con un núcleo cerebral cuya estructura y funciones son poco conocidas: el claustro [9]. El claustro está conectado recíprocamente con todas las regiones de la corteza cerebral, de forma muy significativa con la corteza prefrontal, y su función es muy relevante en el proceso de atención y el estado de conciencia [10,11]. Aunque no se han descrito diferencias significativas en la estructura del claustro en cerebros con TEA [12], sí se han encontrado en la subplaca [13]. El estudio de las posibles alteraciones de la conectividad entre el claustro y la corteza cerebral en el TEA y el TDAH parecen un prometedor proyecto para entender su fisiopatología.
En pacientes con TEA y TDAH se han descrito alteraciones del desarrollo inicial de las sinapsis en los circuitos de conexión entre áreas corticales de procesamiento complejo (que reciben y procesan de forma combinada información multimodal), sobre todo de los lóbulos frontal, temporal y parietal [7,13,14]. El proceso de sinaptogénesis está regulado por múltiples factores genéticos y epigenéticos (ambientales), por lo que corre un alto riesgo de ser alterado en el período perinatal, durante su etapa de mayor maleabilidad, dando como consecuencia trastornos del neurodesarrollo. En relación con el carácter poligénico del TEA se han descubierto genes cuyas mutaciones producen alteraciones sinápticas que cursan con TEA y TDAH, así como discapacidad intelectual y trastornos neuropsiquiátricos. Entre los genes descritos están los que codifican proteínas de organización sináptica, que incluyen complejos de adhesión celular y factores secretados [15]. Muchas proteínas codificadas por genes de riesgo para padecer TEA, TDAH o discapacidad intelectual participan en diferentes procesos de conectividad neuronal en la sinapsis, incluyendo sistemas proteicos relacionados con receptores para neurotransmisores, como el glutamatérgico (p. ej., GRIN2B), el gabérgico (p. ej., GABRA3 y GABRB3) y el glicinérgico (p. ej., GLRA2), pero también en los mecanismos de neuritogénesis (p. ej., CNTN), el establecimiento de las sinapsis (p. ej., cadherinas y protocadherinas), la conducción neural (CNTNAP2) y la permeabilidad de las membranas neuronales a iones (CACNA1, CACNA2D3 y SCN1A) [2,3]. Algunas de estas proteínas están directamente involucradas en la actividad y la formación de las sinapsis, como las neurexinas (NRXN) y las neuroliginas (NLGN). Otras proteínas forman parte de los andamios necesarios para el posicionamiento de moléculas de adhesión celular y receptores de neurotransmisores en la sinapsis, por ejemplo, los genes SHANK (SHANK1, SHANK2 y SHANK3) [3] y los que codifican las proteínas de la familia Rho-GTPasas [16]. Estas proteínas se unen en grandes plataformas moleculares de interacción con receptores de glutamato y actina asociada a proteínas, afectando de forma muy evidente el desarrollo y morfología de las dendritas. Síndromes del neurodesarrollo asociados frecuentemente con la aparición de TEA, como el síndrome X frágil, presentan anomalías importantes en la estatura de las espinas sinápticas en las dendrítas de las neuronas corticales. La distribución heterogénea en intensidad y localización de estas alteraciones en las conexiones neuronales de la corteza explicaría las diversas manifestaciones clínicas, tanto de la entidad diagnóstica (TEA, TDAH, discapacidad intelectual, etc.) como de las diferencias entre individuos con el mismo diagnóstico. EL CANTO DE LAS NEURONAS DISPOSICION DE LAS ESPINAS DENDRITICAS La corteza o córtex cerebral es el tejido nervioso que cubre la superficie de los hemisferios cerebrales, alcanzando su máximo desarrollo en los primates. Es aquí donde ocurre la percepción, la imaginación, el pensamiento, el juicio y la decisión. Es ante todo una delgada capa de la materia gris –normalmente tiene 6 capas de espesor–. Esta capa de células conecta con una amplia colección de vías de materia blanca, que son mucho mas numerosas y son estas preolongaciones que la conectan con todo el resto del cerebro. La delgada capa está depositada en las circunvoluciones, lo que permite el mayor aumento numérico de estas células. Si se extendiese, ocuparía unos 2500 cm². Esta capa incluye unos 10.000 millones de neuronas, con cerca de 50 trillones de sinapsis. La neo corteza o corteza cerebral, localiza una serie de funciones del cerebro que nos hacen ser humanos. Esta localizada en la superficie del cerebro y tiene un espesor de entre uno y cuatro mm se llama sustancia gris y está en contacto íntimo con la sustancia blanca en alusión a sus colores aproximados. En la corteza se depositan las células nerviosas que componen la sustancia gris y la sustancia blanca son las prolongaciones de estas células que están envueltas en mielina a la que deben su color En la corteza del cerebro se alojan las células del sistema nervioso en cantidades de miles de millones- Para visualizar esas células hay que teñirlas. Lo que permite verlas individualmente. Estas células de la corteza cerebral se localizan en forma de capas. Con la tinción de las neuronas se vio que tienen un cuerpo llamado soma del que salen prolongaciones. Las aferentes se llaman dendritas y las eferentes cilindroejes. Son como árboles y de hecho la palabra dendrita proviene del griego que significa árbol. Gracias a las tinciones podemos ver estas células y sus prolongaciones, con el método de Nissen, se tiñen los cuerpos neuronales, mientras que con el método de Golgi se tiñen las prolongaciones de la neurona. Con estas técnicas no se tiñen todas la células, y si algunas, pues de lo contrario no veríamos nada a destacar. El teñido de las células nerviosas es selectivo. Las neuronas forman un bosque tan denso, donde todo está en contacto con todo. Las células nerviosas se comportan como un pequeño ordenador. La información llega a través de las dendritas al cuerpo celular o soma, se elabora una respuesta que a través de una prolongación generalmente más larga, el cilindroeje, y son transmitidas a la sinapsis. Aquí se pone en contacto con las dendritas receptoras, pero no directamente, sino a través de un espacio sináptico. A medida que sale información de los cilindroejes se contacta con las células vecinas. En las terminaciones presinapticas, existen unos depósitos de neurotransmisores que el “ los descarga al espacio sinaptico y desde aquí se estimula la neurona postsinaptica, y empieza el proceso de nuevo. Cajal con su entusiasmo y su curiosidad dijo “conocer el cerebro es conocer el camino de nuestro pensamiento y de nuestras capacidades». El estudio del cerebro nos permite conocer su creatividad y nuestras funciones superiores . Nosotros somos nuestro cerebro y nuestra capacidad de hacer. El conocimiento del cerebro no sólo tiene importancia en el desarrollo filosófico y científico, sino porque nos permite conocer la patología que lo afecta con frecuencia y buscar su tratamiento. Antes de conocer un cerebro alterado necesitamos conocer su morfología normal y su función. La ciencia de nuestro tiempo estudian insistentemente el diseño del cerebro. Su estudio nos llevarán al conocimiento de sus circuitos normales y patológicos. Y despues a su función. Las imágenes del universo con sus enormes magnitudes, son superponibles a las imágenes de la microscopía cerebral con sus pequeñas células que se miden por milésimas de micra.
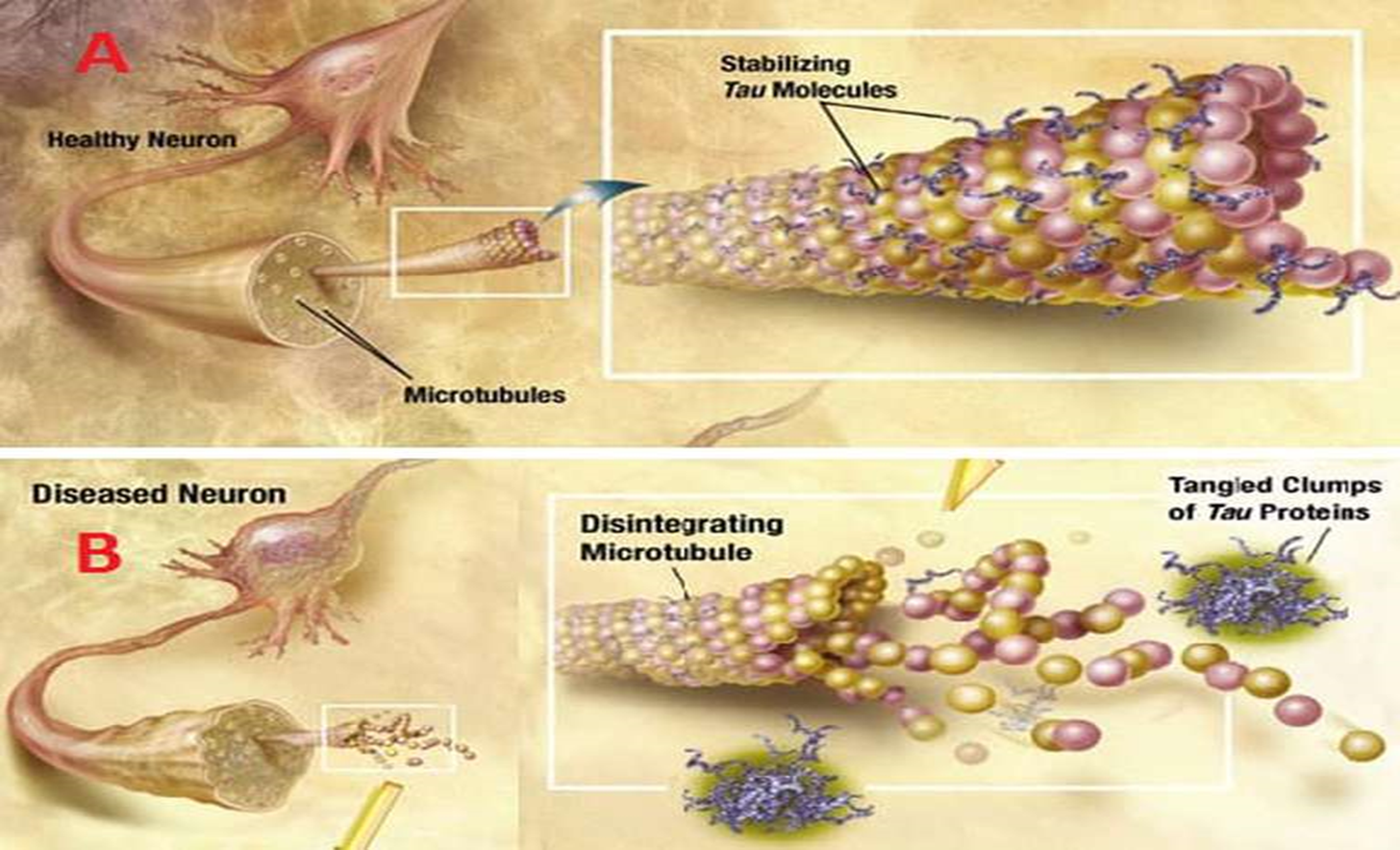
Estamos en un mundo global nadie puede trabajar sólo. Múltiples datos publicados sobre el cerebro, tienen dificultad para entenderlos. Por ello Javier de Felipe forma un grupo heterogeneo de cientificos, que de manera individual, però conjuntandose , intentan entenderlo globalmente. Como modelo de estudio se está utilizando las células piramidales, que se puede decir son las células principales del cerebro. Y son la principal fuente de conecciones. Necesitamos conocer como esta constituido nuestro cerebro para después expresarlo matemáticamente. La razón de este estudio son las espinas dendriticas, evaginaciones del citoplasma de las dendritas, que son las vías aferentes de la neurona. Las células piramidales, se transforman en modelos virtuales a través de la matemática para poder manipularlos con mayor facilidad. Lo que se llama simulación por ordenador. Las espinas dendriticas que parecen una simple prolongación, son estructuras muy complejas que están formadas por más de 500 proteínas y lo importante es que su morfología refleja su función. Las espinas actúan como un pequeño ordenador. Su morfología refleja su función, las que que tienen cabeza grande tienen mayor capacidad de contactar, y cuando se estrechan, la corriente elèctrica, los potenciales de accion, disminuyen. Hay unas espinas de cabeza grande, que tienen gran capacidad de conexión, que son muy estables. La combinación de varios espinas són depósitos de memoria. Otras espìnas, son mas delgadas y son de aprendizaje Una neurona tiene 20.000 espinas. Para ver el detalle de la célula, se introduce un micro electrodo y se inyecta una sustancia fluorescente que permite ver las dendritas y sus prolongaciones. La neurona piramidales son las que tienen mayor número de conexiones dentro del cerebro y son responsables de las funciones cerebrales superiores. En la enfermedad de Alzheimer las espinas dendríticas desaparecen y al ser portadoras de la memoria, se pierde esta. El conocimiento de las espinas dendríticas su formación y su función nos permitirá reparar la pérdida de esta estructura que ocurren en las enfermedades demenciales. Hay millones de neuronas piramidales y como hemos dicho cada una de esta tiene 20.000 espinas. El contajé de las espinas dendríticas no se hace directamente, sino por estimación. Los investigadores han fabricado herramientas que cuentan automáticamente el número de espinas en una dendrita. Hasta ahora el contajé de cada espina costaba meses, mientras que con las herramientas de que se dispone se hace en segundos. El tamaño de las espinas es fundamental para saber la corriente que generan, por lo que hay que calcular el volumen y construirlas en tres dimensiones. Se está intentando obtener los valores de lo volúmenes de forma automática.
La disposición de las espinas en un rosal es similar a la disposición de las espinas dendríticas, aunque las espinas del rosal son distintas pues son puntiagudas y de base ancha. Se intento disponer las espinas del rosal de varias formas espacial y buscar la fórmula matemàtica que expresara su disposición. Los intentos de ver la disposicon en un rosal, fallaron, El contaje y disposicion de las espinas, no se puede expressar matematicamente, ni de forma lineal ni elíptica, por lo que se intenta convertirlas en notas musicales y asi darle una disposicon contable. Los parámetros que componen la disposición, orientación, longitud, forma, disposición de las espines asi como las corrientes de accion, se las convirtio en notas musicales,. Lo que a conseguido avier de Felipe, es considerar notas musicales las distintas espinas dendríticas, de una dendrita, de una células piramidales. Se basa para ello en varios parámetros y así construye una melodía, que sin ser excesivamente precisa nos puede dar un patrón matemático de estas dendritas fundándose en su sonoridad. Un programa matemático donde se convierten en notas musicales un fragmento de dendrita piramidal,los filopodios se han convertido en notas musicales y un arreglo musical, permite obtener una sonoridad bastante clara al interpretarla. Pone una nota a cada espina y una vez arreglada escuchar su melodía. Ya que visualmente no es interpretable. El sonido se repite como un morse, se repite en notas con intervalos de silencio Esto proporciona el sonido de una sola dendrita, y hay que imaginar como suenan todas las espinas de todas las dendritas. Por lo pronto esta técnica es muy limitada pero muy significativa. Y tiene cierta belleza. Estudia grupos de dendrita de un cerebro de 80 años y otro de 40.. En el cerebro de un paciente de 40 años tiene gran riqueza riqueza de notas, porque tiene muchas espines dendriticas. Por el contrario en el cerebro de un paciente con 80 años, las dendritas tienen menos espinas y por tanto menos sónoridad. Con ello se ha construido una neurona virtual, que ha estudiado un ordenador neuromorfico. Cuando se activan determinadas espinas dendríticas, los resultados sonoros son distintos a cuando es estimulan otras distintas. Esto hace que dependiendo de la abundancia de espinas, los estímulos que llegan al cuerpo celular sea diferente. Y por consiguiente los resultados seran diferentes. Una células piramidal es como el conjunto de muchos ordenadores. El problema es cómo utilizar todas estas dendritas, sus espinas e integrarlas en un ordenador. Los ordenadores neuromorfico están adecuados a la faena de contar y analizar espinas , ya que los ordenadores ordinarios está muy lejos de tener esta potencia. Nuestro cerebro al funcionar consume solamente 12 Watios y un superordenador, consume cientos de miles de watios Aplicando este estudio a la enfermedad de alzheimer. Donde se encuentran dos tipos de proteínas anormales. La beta amiloide y la proteína tau y estudiar cómo afecta patológicamente cada una de esta proteínas. El arte nos ayuda también entender la patología de Alzheimer. El ejemplo lo proporciona el pintor William que pintaba autorretratos y que en 1967 empezó a padecer la enfermedad de Alzheimer. Esta enfermedad empieza en el hipocampo, donde se alojaba la memoria inmediata, y desde aquí se expande a otras zonas afectando de forma próxima funciones. Este pintor fue haciendo autorretratos a medida que evolucionaba su enfermedad y es dantesca la deformacion de las imagenes que el va obteniendo de como se ve a si mismo. La enfermedad de alzheimer es progresiva y durante unos xx años permanece con clínica suave con un deterioro cognitivo leve, hasta que el deterioro se hace marcado y muy evidenciable. De forma que morfológicamente se puede ver en los cortes de cerebro de una necròpsia en pacientes con Alzheimer, la extensión y evolución de la enfermedad y relacionarlo claramente con su deterioro cognitivo. Desde que empieza la enfermedad histologicamente, hasta que se manifiesta en su periodo de estado clinicamente. Pasan los años suficientes, como para aplicar la pobre teràpia de que disponemos, , con la posibilidad de que en estos estadios iniciales de la enfermedad, fueran efectivas. Tenemos muchos años para introducir terapias que eviten la evolución del deterioro. El tener marcadores biologicos para esta y su deteccion precoz, permitiria al menos lentificar la enfermedad , en tanto que no aparecen teràpias mas efectivas. La actividad de detereioro socialment invalidante tarda mucho tiempo a partir de las primereras lesiones histologicas en los lobulos temporales. La pregunta es porque determinada afectación cerebral no produce deterioro y si aparece cuando existe una evolución patológica de la lesiones. El estudio se está haciendo inyectando en cerebros normales y patológicos uno marcadores que permiten reconocer como las placas de la enfermedad de alzheimer afectan a las espinas dendríticas. Se trata de ver cómo la placas amiloides afectan a su entorno. Se ha visto claramente que la placa amiloide bloquea claramente las espinas dendríticas y empobrece de espinas las dendritas de estos implantes vitales para la memoria y el aprendizaje. Se ve claramente que alrededor de una placa las espinas desaparecen marcadamente. Las espinas se torna más delgadita y menos numerosas. Las placas mutilan las neuronas y la desconectan del resto. El estudio de cómo actúa la proteína tau se hace con un sistema similar inyectando una sustancia fluorescente que tiñe de rojo el cerebro y permite ver si tiene o no, la tau. Ya que puede ver el deposito patologico de esta proteina. De forma que la proteína TAU no produce disminución de las espinas dendríticas, por lo menos al principio. A partir de aqui Javier de Felipe con la colaboracion de investigadores y sobre todo de musicos, han hecho partituras en preparaciones de pacientes con tau positivo y Tau negativo. La conversión en partitura muestra que los pacientes con tau positivo tiene más silencios y los volúmenes son más pequeños, mientras que la preparaciones sin TAU, son mas sonoras. La música está sirviendo para para obtener datos que no se ven a simple vista Cuando aumenta el depósito de la Tau, se pierden las espinas claramente. Y por tanto se pierden las comunicaciones entre neuronas. Luis Buñuel que también padeció esta enfermedad, tuvo la suficiente claridad de idees, para decir: Hay que haber comenzado a perder la memoria aunque sólo sea retazos, para darse cuenta que la memoria es la que constituye nuestra vida. Nuestra memoria es nuestra coherencia, nuestra razon, nuestros sentimientos, si en ella no somos nada. Un cuarteto intèrpretò la lectura de las música obtenida de las espinas dendríticas. Con un exito extraordinario y puede ser oido en You Tube. La analogía entre el macro cosmo y el microcosmos, tienen tal similitud, que permite la confusión. Multiples pequeños corpusculos en el caso del cerebro, o múltiples inmensos corpusculos en el caso de los planetas que estan enlazados por fibras. Las imagenes, salvando las magnitud, permite la confusion. Deduzco otra vez que existe un patrón de forma en el Universo para lo grande y lo muy pequeño. La similitud enorme de distintos corpusculos, rodeados de halos, parecen los mismos modelos pero a distintas escalas. Cabe una pregunta, si las forma es tan parecida, lo seran tamnbien las funciones. Funcionarà un cerebro como un universo Kepler tambien musicalizo el universo de manera similar a como lo hace de Felipe con las dendritas y aqui añado el relato:
Harmonices mundi ( La armonía de los mundos , 1619 ) es un libro escrito por Johannes Kepler en la ciudad de Linz . El libro contiene la primera formulación de la tercera ley del movimiento planetario . A Harmonices mundi Kepler intenta explicar los movimientos planetarios con base en un modelo geométrico de proporciones entre diferentes poliedros relacionando estos con escalas musicales. En esta obra muestra sus intentos de fijar las órbitas de los planetas en el interior de poliedros perfectos, o sólidos platónicos , tal como había hecho en una obra anterior, misterium Cosmographicum . Para gran decepción suya la teoría nunca funcionó y después de haber expuesto en largas páginas en esta obra la abandona finalmente mostrando que es incompatible con las observaciones y las leyes del movimiento planetario deducidas en Astronomía Nueva . Kepler intentó describir estos movimientos postulando una fuerza similar al magnetismo que él pensaba emanaba del Sol . Kepler expuso en esta obra su teoría de que cada planeta produce un tono musical durante su movimiento de revolución alrededor del Sol y que la frecuencia del tono varía con la velocidad angular de los planetas. Algunos planetas producen notas musicales constantes: por ejemplo la Tierra sólo varía un semitono con una proporción de 16:15 (o equivalentemente la diferencia entre una nota mi y uno hace entre su afelio y su perihelio ) y Venus varía en un intervalo más reducido de 25:24. Kepler explica su razonamiento para deducir el reducido espacio de tonos propio de cada planeta en términos esotéricos. « La Tierra canta Mi, Fa, Mi: se puede deducir de estas sílabas que en nuestro hogar podemos esperar mí seria y hace m. » En momentos muy poco frecuentes todos los planetas podrían tocar juntos en perfecta concordancia. Kepler propuso que esto podría haber pasado una única vez en la historia, quizás en el momento de la creación. En un libro anterior Astronomía nueva , Kepler había escrito las dos primeras leyes del movimiento planetario . La tercera ley, que indica que el cubo de la distancia media del planeta al Sol es proporcional al cuadrado de su período orbital, aparecía por primera vez en el capítulo 5 de este libro después de una larga discusión en astrología .
Bibliografía:
Bedford S.A. et al. Mol Psychiatry 25, 614-628 (2020) PubMed
El Human Brain Proyet, del que Javier de Felipe es el director está dividido en 11 subproyectos. Mas de 150 laboratorios 26 países donde más de 800 sabios intentan , conocer la complejidad del cerebro humano.
[1]Barón-Mendoza I, Maqueda-Martínez E, Martínez-Marcial M, De la Fuente-Granada M, Gómez-Chavarin M, González-Arenas A (2021) Changes in the Number and Morphology of Dendritic Spines in the Hippocampus and Prefrontal Cortex of the C58/J Mouse Model of Autism. Front Cell Neurosci 15: 726501.
Hutsler JJ, Hong H (2010) Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res 1309: 83-94.
Pagani M, Barsotti N, Bertero A, Trakoshis S, Ulysse L, Locarno A, Miseviciute I, De Felice A, Canella C, Supekar K, Galbusera A, Menon V, Tonini R, Deco G, Lombardo MV, Pasqualetti M, Gozzi A (2021) mTOR-related synaptic pathology causes autism spectrum disorder-associated functional hyperconnectivity. Nat Commun 12(1): 6084
El blog de José Ramón Alonso. Neurociencia Exceso de sinapsis en las personas con TEA
Esta claro que este descubrimiento en plena epidemia de Covi, ha sido impactante.
Varias publicaciones en periódicos y revistas medicas informaticas, han aparecido en solo unos días
El nuevo tratamiento de Pfizer, llamado PAXLOVID, consiste en 30 píldoras que se toman a lo largo de cinco días. Combina dos fármacos antivirales.
Por un lado, EL INHIBIDOR DE PROTEASAS PF-07321332, QUE INHIBE UNA ENZIMA QUE EL CORONAVIRUS NECESITA PARA REPLICARSE.
Por otro, EL GENÉRICO RITONAVIR, APROBADO DESDE HACE 25 AÑOS PARA EL TRATAMIENTO DEL VIH, QUE FRENA LA DEGRADACIÓN DEL PF-07321332 EN EL HÍGADO, Y PERMITE ASÍ QUE SE MANTENGA ACTIVO DURANTE MÁS TIEMPO.
Si esto lo confirma la practica es revolucionario y además admite la pregunta “y ahora con la vacunas que”
Iniciar el tratamiento en los cinco primeros días de síntomas reduce drásticamente las hospitalizaciones y las muertes
Fábrica de producción de Pfizer en Puurs (Bélgica)
Una píldora antiviral de Pfizer reduce el riesgo de hospitalización por covid en un 85% si se empieza a tomar en los cinco primeros días desde el inicio de síntomas, según resultados de un ensayo clínico realizado en personas con alto riesgo de sufrir complicaciones graves.
El tratamiento es el segundo de una nueva generación de fármacos contra el SARS-CoV-2 que se toman por vía oral y que abren la vía a iniciar la terapia antes de que la enfermedad se agrave. Sus resultados llegan pocas semanas después que los del molnupiravir de Merck & Co, aprobado el jueves en el Reino Unido, pero aún no en la Unión Europea ni en Estados Unidos.
Hasta ahora, los únicos tratamientos antivirales disponibles contra el SARS-CoV-2 -el fármaco remdesivir y los anticuerpos monoclonales- se administran por vía endovenosa, lo que limita su uso al ámbito hospitalario.
El nuevo tratamiento de Pfizer, llamado PAXLOVID, consiste en 30 píldoras que se toman a lo largo de cinco días. Combina dos fármacos antivirales.
Por un lado, EL INHIBIDOR DE PROTEASAS PF-07321332, QUE INHIBE UNA ENZIMA QUE EL CORONAVIRUS NECESITA PARA REPLICARSE.
Por otro, EL GENÉRICO RITONAVIR, APROBADO DESDE HACE 25 AÑOS PARA EL TRATAMIENTO DEL VIH, QUE FRENA LA DEGRADACIÓN DEL PF-07321332 EN EL HÍGADO, Y PERMITE ASÍ QUE SE MANTENGA ACTIVO DURANTE MÁS TIEMPO.
Según los resultados anunciados por Pfizer en un comunicado, de las 607 personas que recibieron el tratamiento en un ensayo clínico, seis fueron hospitalizadas en los 28 días siguientes (un 1%) y ninguna murió. Entre las 612 que recibieron un placebo, 41 fueron hospitalizadas (6,7%) y diez murieron.
El sistema es muy sencillo y facilita la medicación antiviral, tan escasa de forma que además esta medicación es alentadora por incrementar los antivuirales tan escasos
Estos datos implican que el paxlovid ha mostrado una eficacia del 85% para prevenir hospitalizaciones cuando el tratamiento empieza en los cinco primeros días desde el inicio de síntomas. Si se limita el análisis estadístico a casos que empiezan el tratamiento en los tres primeros días, la eficacia sube al 89%.
Con estos resultados, haría falta tratar a 18 pacientes con paxlovid para evitar una hospitalización. La cifra es similar a la obtenida con el molnupiravir de Merck, con el que haría falta tratar a 15 pacientes para prevenir una hospitalización, según datos aportados por Roger Paredes, jefe de sección de enfermedades infecciosas del hospital de Can Ruti en Badalona, que lidera un equipo que ha participado en el desarrollo clínico de ambos antivirales.
Hará falta tratar a 18 pacientes de alto riesgo para evitar una hospitalización
Aunque la eficacia del 85% antiviral de Pfizer es aparentemente más alta que la del de Merck, que redujo el riesgo de hospitalización en un 50%, es una apariencia engañosa porque «los ensayos clínicos se realizaron en poblaciones distintas, por lo que los resultados no son comparables», advierte Paredes.
Esto parece una competencia
En el caso del molnupiravir, un 14,1% de los pacientes tratados con placebo fueron hospitalizados, frente al 6,7% registrado en el ensayo clínico del paxlovid. Esto indica que el molnupiravir se ensayó en una población con más riesgo de complicaciones graves. Según el investigador de Can Ruti, «lo que nos permite comparar mejor la eficacia de estos fármacos es analizar el número de personas que hay que tratar para evitar una hospitalización».
La eficacia del paxlovid de Pfizer para prevenir hospitalizaciones es similar a la del molnupiravir de Merck
Los resultados del antiviral de Pfizer, «son muy buenos en población de alto riesgo no vacunada», destaca Paredes. «Cuando este fármaco esté aprobado, ayudará a evitar ingresos por covid. Pero falta definir cuáles son los colectivos de alto riesgo en los que estará indicado en poblaciones con tasas altas de vacunación».
El ensayo clínico se ha realizado entre julio y septiembre en América, Europa, Asia y África, cuando la variante delta del coronavirus ya era dominante. Aunque el plan inicial preveía incluir a 3.000 pacientes en el ensayo, se ha tomado la decisión de interrumpirlo antes de llegar a esta cifra al haberse demostrado ya la elevada eficacia del tratamiento.
Los posibles efectos secundarios han sido similares entre las personas que han recibido el antiviral de Pfizer que entre los que que han recibido placebo. Concretamente, un 19% de las personas tratadas con Paxlovid y un 21% de las que han tomado un placebo han notificado posibles efectos adversos, la mayoría de ellos leves.
Pfizer ha anunciado hoy que presentará los datos del ensayo clínico a la Agencia de Alimentos y Fármacos de EE.UU. (FDA) para solicitar la autorización del paxlovid lo antes posible. La compañía no ha informado de sus planes para obtener la autorización de la Agencia Europea de Medicamentos (EMA) para distribuir la píldora antiviral en la Unión Europea.
Los efectos secundarios no han sido mayores entre las personas que han recibido el fármaco que entre las que han tomado un placebo
«Ya nos hemos asegurado millones de dosis», ha anunciado el presidente de EE.UU., Joe Biden, en declaraciones recogidas por Reuters. «Si la FDA lo autoriza, pronto podemos tener píldoras que tratan la covid en aquellos que se infectan. (…) La terapia sería una herramienta más para proteger a las personas de las peores consecuencias de la covid».
En el ensayo clínico del que ha informado hoy Pfizer solo han participado voluntarios con alto riesgo de sufrir complicaciones graves por la covid. La compañía tiene actualmente en curso otro ensayo clínico, del que aún no hay resultados, para evaluar la eficacia del tratamiento en personas sanas con riesgo bajo de sufrir complicaciones graves por la covid.
Son resultados muy buenos en población de alto riesgo no vacunada. Falta definir cuáles son los colectivos en los que estará indicado este fármaco»
Este señor con cara de buena persona es R David Wallace,físico teórico en Oxford y en la USC; publica ‘The emergent multiverse’ Tiene 44 años dedicados a estudiar el espacio-tiempo desde que conocío ni mas ni menos a Hawking a los 15. Investigué en Oxford durante 20 años y, hoy, en California. El universo no es un solo “verso”, sino multisuperposiciones a nivel micro, cuántico, que no vemos a nivel macro. Diserto en el congreso de ontología en el CCCB
Porque copioteo estas entrevistas, pues no lo se muy bien, pero me gusta y creo que al transcribirla, las puedo entender un poquito mejor. Por si acaso pido perdón al que sea
Un ‘omniverso’emergente
Hemos evolucionado para interpretar lo que vemos a nivel macro, donde un gato no puede estar vivo y muerto a la vez, pero en la realidad cuántica, desconocida pero tan cierta como la que observamos, el gato puede estar vivo y muerto a la vez, al quedarse en superposición entre dos estados de la materia. Fenómenos como ese y otros que aún investigamos –dice Wallace citando a Everett– hacen que funcionen los chips que mueven el mundo en aparatos como el móvil que, paradójicamente, sí parecen reales. Y sostiene que el universo no es un solo “verso”, sino que “pedazos” ( chunk s) de realidad transcurren –sin interferirse– en decoherencia cuántica en un multiverso emergente. La construcción del primer ordenador cuántico –y en Barcelona, el del BSC– podrá descubrirnos quizá cómo pueden suceder dos cosas a la vez.
Usted sostiene que no existe un universo sino muchos…
Un multiverso , sí. Es muy posible que esto sea cierto pero con la cabeza que yo tengo me es absolutamente imposible entenderlo. Solo puedo creérmelo y partir de ahí que Dios reparta suerte.
Hace solo unos días unos experimentadores en un pueblo cercano a Barcelona han demostrado que una partícula podría estar en dos partes al mismo tiempo durante fracciones de segundo. Esto es experimental no es un sueño pero a mi cerebro habituado durante muchos años, a que una cosa solamente puede ser una y está en un mismo sitio, esto le cuesta un tormento.
Tengo entonces, que comprometerme a admitir que detrás del último puede haber alguien.
El mundo que percibo es mi mundo pero no el real, no el verdadero, porque el verdadero no existe es ficción.
Necesito apoyarme en contradiciones, una cosa no puede ser y no ser al mismo tiempo. Pues si lo puede ser, dependiendo del universo donde se aloje.
De forma que de manera elemental, llego a la conclusión de que todo puede ser mentira con la mente que estoy usando o decir
“dilo tu primero”
La historia nos demuestra que ciertas cosas hoy rutinaria condujo a ciertos sabios incluso a la muerte.
Si la tierra daba vueltas alrededor del Sol o el sol alrededor de la tierra condujo al pobre de Galileo a la prisión durante mucho tiempo.
Si no me parece entendible lo que dices, pues te condeno y te quito de enmedio
¿Puede demostrarlo?
Está demostrado, pero nos cuesta aceptarlo, incluso a los científicos.
¿Por qué?
¿Por qué le costó tantos siglos a la humanidad aceptar que la Tierra es redonda y gira alrededor del sol?
Porque no lo parece.
Pero ¿cómo sería la Tierra si sí pareciera que gira alrededor del sol?”
Supongo que como es.
¿Y cómo sería la Tierra si sí pareciera que el gato de Schrödinger está vivo y muerto a la vez como está a nivel cuántico?
También como es ahora, imagino.
Bien. ¿Y cómo sería el mundo si también sí pareciera que vivimos en varios universos a la vez y no en uno solo?
PUBLICIDAD
También como es ahora.
Eso creo yo. Las mismas preguntas se hizo Wittgenstein, y es que, como no parece ser posible que un gato esté vivo o muerto a la vez y, por tanto, varios universos transcurran al mismo tiempo, nos cuesta aceptarlo, aunque la evidencia científica lo demuestre.
¿Cuál es esa evidencia del multiverso ?
Cientos de aparatos hoy demuestran que a nivel cuántico no hay un solo universo.
¿Cuáles?
Su móvil funciona por esa mecánica cuántica que nos dice que no vivimos en un universo, sino en un multiverso . Y lo mismo demuestra cualquier chip de cualquier aparato…
¿Cómo?
Funcionan por las mismas leyes cuánticas que hacen que el gato de Schrödinger pueda estar vivo y muerto a la vez en una caja, según el universo que decide observar el observador al abrirla en su experimento.
¿Cómo puede estar vivo y muerto?
Aún no sabemos cómo, pero sí que la cuántica –las ecuaciones– lo demuestran, porque la realidad es cuántica. Por eso, no solo hay un universo, sino un multiverso . Y en uno el gato sigue vivo y en otro está muerto… El devenir se bifurca, según decida el observador.
¿Y no sigue siendo inexplicable?
Que una partícula puede estar en dos sitios a la vez se llama superposición y esa superposición en este caso macroscópica de un evento microscópico, cuántico, ha sido demostrada, pero si la mides, verás que es inexplicable… Los experimentos nos llevan una y otra vez al gato vivo y muerto.
¿No será un problema de medición?
La cuántica es muy sólida matemáticamente: las ecuaciones dicen que está vivo y muerto en una superposición.
¿Cómo?
El mundo, en resumen, hace al mismo tiempo dos cosas ordinarias y eso es lo que nos parece imposible, como parecía a los antiguos que la Tierra gira alrededor del Sol. En el mundo microscópico de la mecánica cuántica, en cambio, hay dos mundos a la vez: en uno el gato está muerto, y en otro, vivo.
¿Y no coinciden?
Los dos mundos no se comunican; no interfieren uno con otro; solo se superponen a ese nivel de partículas, cuántico; pero en el mundo macro eso no es apreciable. Así que podemos decir que hay dos mundos a la vez: el del gato vivo y el del gato muerto. Y es lo que nos dice la cuántica: que hay muchos mundos.
¿Y podemos llamarle multiverso ?
Yo lo llamo un omniverso emergente. Eso explicaría la cuántica mejor que modificar las ecuaciones porque no parecen reales.
¿El mundo estaría hecho entonces de superposiciones cuánticas como la del gato?
Lo que sostengo es que la estructura fundamental del universo no es un solo verso , una sola dirección, como nos parece, sino que a nivel microscópico cuántico consta de múltiples superposiciones .
¿Eso qué explicaría?
No tenemos aún una teoría del todo para explicar el mundo. Todas las teorías son parciales, pero, por ahora. las más profundas son las cuánticas. El intento que ha llegado más lejos, la teoría de cuerdas, es también cuántico y también consta de superposiciones e interferencia, ramificaciones, decoherencia …, compatibles con la de los multiversos .
¿Qué espera que suceda ahora en el terreno de lo demostrable?
El ejemplo más importante de lo que digo es la construcción de un ordenador cuántico.
¿Por qué?
Porque es el intento de aplicar las superposiciones cuánticas para acelerar los procesos de computación hasta niveles que ningún ordenador ha alcanzado.
¿Y eso demostraría los multiversos ?
Todo lo que nos conduce al ordenador cuántico es a pequeña escala una confirmación de que superposiciones como las del gato de Schrödinger son posibles a todas las escalas. Si su construcción no falla en algún punto, tenemos cada vez más razones para creer en un multiverso .
No dudo que un ordenador cuantico dira mas cosas, pero se las tienen que decir a un cerebro que las entienda, aunque ya existe ese cerebro, por ejemplo el del autor de este trabajo, David Wallace,físico teórico en Oxford y en la USC; publica ‘The emergent multiverse’.
No obtante para desenvolvernos en este campo del multiversos con la misma facilidad que lo hacemos en el mundo de Newton, nos va a costar, es posible que incluso tengamos que cambiar de cerebro
En mi caso me las tienen que decir varias o muchas veces, para que pueda por lo menos asentir con la cabeza, pero no entenderlas
LLUÍS AMIGUET 06/11/2021 00:25Actualizado a 06/11/2021 08:59
Está claro que el coronavirus ha potenciado la investigación en el mundo entero, no solo una serie importante de vacunas sino ahora un antiviral
El nuevo MONULPIRAVIR ofrece unas posibilidades de curación para esta epidemia que no teníamos y que nos alegra muchísimo a todos.
La farmacéutica Pfizer ha confirmado los resultados óptimos de su píldora antiviral PAXLOVID, que reduce el riesgo de hospitalización y muerte por coronavirus hasta un 90%. Además, los responsables de la compañía destacan los «signos positivos» de estos medicamentos contra las diferentes variantes del virus, como la Delta. En un contexto marcado, por la aprobación del primer fármaco oral contra la COVID de la compañía Merck, monulpiravir, para su uso de emergencia, cuya eficacia es de un 50%.
El antiviral, de acuerdo a los datos del ensayo de fase 3, redujo significativamente el riesgo de hospitalización o muerte en casos leves o moderados. Ya se autorizó en Reino Unido y espera la aprobación de la FDA.
El molnupiravir se puede administrar a los pacientes en sus casas, a diferencia del antiviral remdesivir de Gilead Sciences Inc. y otras terapias con anticuerpos monoclonales, que se administran mediante una infusión intravenosa generalmente en un hospital o una clínica. |
Molnupiravir podría convertirse en el primer medicamento para tratar el Covid-19 y evitar las hospitalizaciones y muertes en un 50 por ciento de los casos. Se trata de una pastilla antiviral que se administra por vía oral, e inhibe la replicación del virus.
Según informaron la farmacéutica Merck y su socio Ridgeback Biotherapeutics LP, fabricantes del medicamento, este antiviral redujo significativamente el riesgo de hospitalización o muerte en casos leves o moderados, tras conocerse los datos de un análisis intermedio del ensayo de fase 3.
“En el análisis intermedio, el 7,3 por ciento de los pacientes que recibieron molnupiravir fueron hospitalizados hasta el día 29, en comparación con el 14,1 por ciento de los pacientes tratados con placebo que fueron hospitalizados o fallecieron”, explicó Merck en un comunicado, y detalló además que “se redujo el riesgo de hospitalización o muerte en aproximadamente un 50%”.
En cuanto a los efectos adversos de la medicación, anunciaron que la incidencia fue comparable en los grupos de molnupiravir y placebo (35% y 40%, respectivamente). De manera similar, la incidencia de eventos adversos relacionados con el fármaco también fue comparable (12% y 11%, respectivamente).
De acuerdo a los datos del ensayo, del que participaron 775 personas, la eficacia del fármaco «no se vio afectada por el momento de aparición de los síntomas o el factor de riesgo subyacente». Además, según los participantes con datos de secuenciación viral disponibles (aproximadamente el 40% de los participantes) «el molnupiravir demostró una eficacia constante en las variantes virales Gamma, Delta y Mu».
El molnupiravir, se puede administrar a los pacientes en sus casas, a diferencia por ejemplo del antiviral remdesivir de Gilead Sciences y otras terapias con anticuerpos monoclonales, que se administran mediante una infusión intravenosa generalmente en un hospital o una clínica. Tratar a los pacientes con covid-19 en casa evitaría riesgos asociados a la transmisión del virus al personal médico y a otros pacientes.
De acuerdo a información del New York Times, un tratamiento de cinco días de molnupiravir tendría un costo de alrededor de US$700 por paciente, un tercio de lo que cuesta un tratamiento con anticuerpos monoclonales.
Tras este anuncio, la mayor distribuidora de vacunas contra la COVID en la Unión Europea ha confirmado que enviará la documentación del estudio en el menor tiempo posible a la Agencia de Alimentos y Medicamentos de Estados Unidos (FDA) para su aprobación y su uso de emergencia en pacientes infectados por el virus.
El estudio de la farmacéutica indica que «los pacientes que tomaron el medicamento con otro antiviral tuvieron una reducción de hospitalización y deceso del 89%», en comparación de aquellos que tomaron un único placebo. «Este antiviral oral tiene el potencial de salvar la vida de los pacientes y elimina hasta nueve de cada diez hospitalizaciones», ha ratificado Albert Bourla, director ejecutivo de Pfizer.
El laboratorio ha especificado que el estudio realizado se basa en 1.219 adultos que contaban al menos con una afección médica y una infección vírica confirmada por las pruebas de detección de la COVID. También, recibieron una dosis baja de ritonavir, un fármaco que se usa en tratamientos contra el sida.
Además, la compañía estadounidense ha detallado que, de los 607 participantes, solo hubo seis hospitalizaciones y cero muertes en pacientes con una alta carga viral, a diferencia de las 612 personas que tomaron un placebo, con un total de 42 ingresados y diez muertes por coronavirus.
Pfizer podría empezar su distribución a nivel mundial durante este año, en el caso de que obtenga la autorización por parte de la FDA.
Una vez aprobada, este fármaco oral se utilizará en pacientes infectados o para prevenir más contagios dentro de un núcleo familiar o empresarial.
Esperamos todas las venturas para esta medicación y sus pacientes
Este trabajo comenta que todo lo que sucede tiene su causa, pero no siempre.
Es conocido por todos que en una epidemia, sea la que sea, no enferma todo el mundo.
Si que hay epidemias, que afectan a mas personas que otras, y la pregunta popular es:
Que pasa porque unos se infectan y otros no.
Que duda clave que la genética tienen un papel, determinadas personas con alteraciones genómicas van a enfermar, y no lo harán los que no sufren estas mutaciones.
¿Con las bendiciones que traería la confirmación del trabajo de Jean Laurent Casanova.¿
Se puede localizar por el estudio del genoma, las personas que afectadas por una mutacion en sus cromosomas están mas expuestos a múltiples enfermedades, que aparecerían cuando el medio les sea propicio
Y mayor bendición, cuando la tecnología sea capaz de reparar estas mutaciones antes que enfermen
Podemos saber desde que nacemos por las alteraciones genéticas a qué enfermedades graves estamos expuestos
Multiples personas se infectan de virus, bacterias u hongos sin sufrir las patologías graves que sí causan en una minoría de los infectados.
Si dispusiéramos de nuestro mapa genéticos podríamos anticipar diagnósticos y tratamientos.
La inteligencia artificial nos puede ayudar a explicar por qué la genética de unos se convierte en mortal mientras que esta ,misma infección es inocua en otros.
Un mismo virus, bacteria u hongo puede infectar a millones de personas y resultar inofensivo para la mayoría, pero muy dañino, incluso mortal, para una pequeña proporción de infectados.
¿Porque ocurre esto.
La variación entre individuos es enorme.
Porque unos pacientes de covid, en principio igual de sanos que el resto de la población, han sufrido un cuadro grave o han muerto, mientras los demás apenas padecían.
¿Esto solo se puede explicar por la conjunción entre los agentes infecciosos y la genética de cada paciente.
Algunos genotipos predisponen a que una infección se manifieste de forma grave.
¿Intentar buscar un mecanismo que explique esta conjunción seria de gran valor.
Sabemos que determinadas enfermedades eran producidas por gérmenes, sin que se hubiera pensado en ello.
La úlcera de estómago es causada por bacterias y los descubridores de la helicobacter pylori ganaron el Nobel por ello.
Otro ejemplo es la enfermedad de Whipple, también causada por una bacteria que afecta tan solo a 1 de cada millón de personas. En España, con 50 millones de habitantes, solo la sufren 50 pacientes.
Es imprescindible explicar que ocurre para que solo algunos pacientes se contagien. Se sabe qué una bacteria que infecta al 20% de los españoles –10 millones de personas– solo acaba enfermando a 50. Y ello se debe a la mutacion del gen IRFKA. Así que el patrimonio genético de cada uno es determinante. Y creemos que pasa igual con muchas otras enfermedades. Y esto podría ser aplicado a la Covid.
Llegados este punto es fundamental saber que individuos estas predispuesto a sufrirla de forma grave?
La secuenciación del genoma de cada individuo, permitiría conocer su alteraciones genómicas y después buscar la adecuada para que no se desarrolle la enfermedad.
Seguro que esto se conseguira, porque lo difícil ya esta hecho y es que se la secuenciación del genoma hoy ya nos permite predecir el riesgo de cáncer de mama vía mutaciones de BRCA1 o el de enfermedad de Parkinson por mutaciones en el gen parkinson.
¿El genoma secuenciado diría a qué enfermedades estamos predispuestos?
Así es y, por tanto, sería mejor realizar la secuenciación al nacer o de niños.
¿Y el alzheimer? ¿Podríamos saber nuestra predisposición con la secuenciación?
No tengo datos al respecto.
¿Puede generalizarse ese esquema?
Creo que existe un esquema general, en efecto, de las enfermedades infecciosas severas, dejando de lado cuál sea el agente infeccioso. No es que unos sufran más una enfermedad infecciosa por coincidencia o por mala suerte…, es su genoma el que lo determina.
¿Podríamos anticiparnos y saber quiénes están predispuestos antes de contraerla?
Con medicina genética personalizada. Y un buen ejemplo es la covid, de la que entendemos solo un 20% de los casos. Y nos gustaría entender más, sobre todo los más severos.
¿Qué esperan encontrar en ellos?
Con el estudio de esos casos y de otras enfermedades infecciosas, como la tuberculosis, intentamos establecer una teoría general con un esquema para definir la arquitectura genética e inmunológica humana en enfermedades infecciosas fatales.
¿Qué ha aprendido de esta pandemia?
Que la deficiencia de interferón tipo 1 subyace en un 15%-20% de casos críticos.
¿Es relevante para futuras pandemias?
Describe un mecanismo general para la neumonía fatal por covid, porque los pacientes con una deficiencia detectable de interferón tipo 1 son, en cambio, clínicamente no diferenciables de los demás, sea cual sea la causa.
¿Serviría para describir otras?
Creo que si entendemos los determinantes de otras formas de infección en otros SARS, neurocovid, covid de larga duración…, entre todos completaríamos esa teoría general de las enfermedades infecciosas.
¿Quiénes son todos?
Formamos el consorcio Covid Human Genetic Effort, en el que destacan los grupos españoles y, aunque estemos ahora en Madrid, quiero subrayar que los equipos catalanes son muy productivos, como verá en nuestra web, porque los hospitales e instituciones de Catalunya de probada capacidad se han implicado en nuestro esfuerzo.
Los vasos linfáticos están distribuidos por todo nuestro cuerpo, pero hasta el momento había poca evidencia que sugiriese que el cerebro también los contiene.
Una innovadora investigación de los Institutos Nacionales de Salud muestra que el cerebro tiene vasos linfáticos, lo que le permite procesar «residuos» que se filtran de los vasos sanguíneos. Este hallazgo arroja luz sobre la relación entre el cerebro y el sistema inmunológico.
Estudios recientes confirman que el cerebro también cuenta con tubos de drenaje para procesar los residuos.
El sistema linfático del cuerpo depende de los vasos linfáticos para absorber, procesar y devolver las proteínas y el líquido tisular, es decir, el líquido que rodea las células del tejido del cuerpo al torrente sanguíneo. El líquido transportado por los vasos linfáticos se llama «linfa».
Hay tres funciones principales del sistema linfático: ayudar a mantener constante el volumen y la presión sanguínea, luchar contra agentes externos como parte del sistema inmunológico y absorber las vitaminas liposolubles y grasas.
Un estudio sobre ratones en 2015 mostró que los vasos linfáticos estaban presentes en el sistema nervioso central de los roedores, lo que sugería que esto mismo podría ser cierto para los seres humanos y otros primates.
Un equipo de investigadores de los Institutos Nacionales de Salud, dirigido por el científico Daniel S. Reich, ha confirmado que los cerebros humanos también tienen vasos linfáticos; en concreto, en la duramadre. «Vimos cómo los cerebros de las personas drenaban fluidos a estos vasos”. Los investigadores utilizaron métodos no invasivos para determinar la presencia de vasos linfáticos en el cerebro humano, la resonancia magnética
Cinco voluntarios sanos y tres monos tití cuyos cerebros fueron escaneados usando resonancia magnética, después de inyecciones con diferentes tipos de agentes de contraste, que permite a los científicos visualizar mejor los vasos sanguíneos en el cerebro.
Primero inyectaron gadobutrol, cuyas moléculas son lo suficientemente pequeñas como para salir de los vasos sanguíneos hacia el compartimento intersticial del cerebro. La exploración por resonancia magnética reveló líneas delgadas más resaltadas, esto es, los vasos linfáticos que habían recogido el agente de contraste que se había escapado de los vasos sanguíneos.
Según los científicos, hay vasos linfáticos en la duramadre que recogen elementos fluidos del compartimiento intersticial del cerebro y al igual que otros órganos en el cuerpo, el fluido cerebral puede drenar a través del sistema linfático
Los autores del estudio, que publica la revista Life, esperan investigar ahora qué implicaciones tienen estas conclusiones desde una perspectiva clínica. Por ejemplo, les gustaría ver cómo funciona el sistema linfático en las personas diagnosticadas con enfermedades neuroinflamatorias como la esclerosis múltiple.
La existencia de nódulos semejantes a los ganglios linfáticos en el cerebro de pacientes con cáncer cerebral pueden ayudar a combatirlos
Investigadores de la Universidad de Uppsala, en Suecia, sugiere nuevas oportunidades para regular la respuesta antitumoral del sistema inmunitario
Confirman que el cerebro también cuenta con tubos de drenaje para procesar los residuos.
De igual forma una investigación de los Institutos Nacionales de Salud muestra que el cerebro tiene vasos linfáticos, lo que le permite procesar «residuos» que se filtran de los vasos sanguíneos.
Este hallazgo arroja luz sobre la relación entre el cerebro y el sistema inmunológico. El sistema linfático del cuerpo depende de los vasos linfáticos para absorber, procesar y devolver las proteínas y el líquido tisular, es decir, el líquido que rodea las células del tejido del cuerpo al torrente sanguíneo transportado por los vasos linfáticos se llama «linfa».
Hay tres funciones principales del sistema linfático: ayudar a mantener constante el volumen y la presión sanguínea, luchar contra agentes externos como parte del sistema inmunológico y absorber las vitaminas liposolubles y grasas.
Un estudio sobre ratones en 2015 mostró que los vasos linfáticos estaban presentes en el sistema nervioso central de los roedores, lo que sugería que esto mismo podría ser cierto para los seres humanos y otros primates. Ahora, un equipo de investigadores de los Institutos Nacionales de Salud, dirigido por el científico Daniel S. Reich, ha confirmado que los cerebros humanos también tienen vasos linfáticos; en concreto, en la duramadre, o la membrana externa más gruesa que rodea al cerebro. «Literalmente vimos cómo los cerebros de las personas drenaban fluidos a estos vasos. Gracias a la resonancia magnética Los investigadores utilizaron métodos no invasivos para determinar la presencia de vasos linfáticos en el cerebro humano.
Trabajaron con cinco voluntarios sanos y tres monos tití cuyos cerebros fueron escaneados usando resonancia magnética, después de inyecciones con diferentes tipos de agentes de contraste, que permite a los científicos visualizar mejor los vasos sanguíneos en el cerebro. Primero inyectaron gadobutrol, cuyas moléculas son lo suficientemente pequeñas como para salir de los vasos sanguíneos hacia el compartimento intersticial del cerebro. La exploración por resonancia magnética reveló líneas delgadas más resaltadas, esto es, los vasos linfáticos que habían recogido el agente de contraste que se había escapado de los vasos sanguíneos.
Según los científicos, hay vasos linfáticos en la duramadre que recogen elementos fluidos del compartimiento intersticial del cerebro. Podemos ver finalmente que, al igual que otros órganos en el cuerpo, el fluido cerebral puede drenar a través del sistema linfático».
Los autores del estudio, que publica la revista eLife, esperan investigar ahora qué implicaciones tienen estas conclusiones desde una perspectiva clínica. Por ejemplo, les gustaría ver cómo funciona el sistema linfático en las personas diagnosticadas con enfermedades neuroinflamatorias como la esclerosis múltiple.
«Estos resultados podrían cambiar fundamentalmente nuestra forma de pensar acerca de cómo el cerebro y el sistema inmunológico se relacionan»,
Recientemente otro trabajo de la Universidad de Uppsala, en Suecia, ha descubierto en pacientes con cáncer cerebral unas estructuras similares a los ganglios linfáticos, donde las células inmunitarias pueden activarse para atacar el tumor.
De igual forma han demostrado que la inmunoterapia potencia la formación de estas estructuras en un modelo de ratón.
Este descubrimiento, publicado en la revista Nature Communicacion, sugiere nuevas oportunidades para regular la respuesta antitumoral del sistema inmunitario.
El glioma es un tumor cerebral mortal con un mal pronóstico. Una de las razones por las que los tumores cerebrales son muy difíciles de tratar es que nuestro sistema inmunitario, diseñado para detectar y destruir células extrañas, incluidas las cancerosas, no puede llegar fácilmente al lugar del tumor debido a las barreras que rodean el cerebro.
Los investigadores han descubierto estructuras similares a los ganglios linfáticos en el cerebro donde los linfocitos T podrían activarse
Para luchar contra un tumor en desarrollo, las células inmunitarias asesinas, como los linfocitos T, deben activarse y prepararse en nuestros ganglios linfáticos, antes de desplazarse al lugar del tumor para eliminar eficazmente las células cancerosas. Debido a las barreras que rodean el cerebro, es un proceso difícil para los linfocitos T llegar al tumor.
En el estudio los investigadores describen su descubrimiento de estructuras similares a los ganglios linfáticos en el cerebro donde los linfocitos T podrían activarse. en pacientes con glioma»,
«Estas estructuras se conocen como estructuras linfoides terciarias (TLS) y no se encuentran en individuos sanos -prosigue-. Tienen todos los componentes necesarios para favorecer la activación de los linfocitos in situ, lo que significa que podrían tener un efecto positivo en la respuesta inmunitaria antitumoral».
El alfaCD40 se está probando actualmente para tratar los tumores cerebrales en una serie de ensayos clínicos
Los investigadores también demostraron que la formación de TLS en el cerebro puede ser inducida por un tipo de inmunoterapia en ratones portadores de gliomas. En efecto, cuando trataron a los ratones con anticuerpos inmuno estimuladores denominados alfaCD40, la formación de TLS se vio potenciada y se produjo siempre en la proximidad de los tumores.
«Hace falta aprender como las inmunoterapias pueden modular la formación de estructuras linfoides terciarias en el cerebro y ofrecer oportunidades para encontrar nuevas formas de regular la respuesta inmunitaria antitumoral en el glioma»,.
El alfaCD40 se está probando actualmente para tratar los tumores cerebrales en una serie de ensayos clínicos. En el estudio que ahora se publica, los investigadores descubrieron que, si bien el alfaCD40 potenciaba la formación de TLS, también inhibía de forma contraproducente la capacidad de los linfocitos T de matar tumores. Por lo tanto, el estudio ha aportado importantes conocimientos sobre los efectos multifacéticos de la terapia con alfaCD40.
Referencia:
Martina Absinta et al. Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by MRI, eLife (2017). DOI: 10.7554/eLife.29738
EN LOS GLIOMAS DONDE ESTA AUMENTADA LA CANTIDAD DE PROTEÍNA TAU, DISMINUYE LA NEOANGIOGENESIS
Que relación puede tener un glioma con la enfermedad de Alzheimer, y como la proteína TAU, reparadora de los tubulos conductores de los cilondroejes es además macrofago
En diferentes estadíos del desarrollo neuronal se observan distintas concentraciones de isoformas de la proteína Tau y se ha demostrado que alteraciones en sus proporciones relativas están relacionadas con algunas enfermedades neurodenegerativas como el Alzheimer, Parkisonismo o la Demencia fronto-temporal.
“Utilizando el modelo de glioblastoma y entendiendo el papel de Tau en esta enfermedad y cómo es capaz de regular los vasos sanguíneos podríamos comprender también cuál es el papel de esta proteína en la disfunción neurovascular de enfermedades como el alzhéimer”,
Las neuronas tienen en general una forma muy polarizada con un cuerpo (o soma), dendritas y una larga prolongación llamada axón. En el soma se sintetizan las proteínas, que después son trasladadas al axón para poder ser usadas en la transmisión del impulso nervioso. Para llegar, se desplazan sobre un andamiaje constituído por unas estructuras tubulares que forman una especie de vía, denominadas microtúbulos, por la que se mueve el transporte neuronal. La función de la proteína Tau está relacionada justamente con su unión a los microtúbulos, debido a que el pegado contribuye a la estabilidad y permite el movimiento de cargas sobre ellos, ya sea desde el soma hacia el axón o viceversa.
“Las neuronas producen varias formas de Tau, especialmente dos llamadas 3R y 4R. Durante el desarrollo hay niveles muy altos de 3R y muy bajos de 4R, pero que alcanzan una proporción de 50 y 50 por ciento en el cerebro adulto”, explica Tomás Falzone, investigador adjunto del CONICET en el Instituto de Biología Celular y Neurociencia (IBCN, CONICET) y coordinador del trabajo.
Sin embargo, hay un desbalance en esas proporciones en ciertas enfermedades neurodegenerativas. “Por ejemplo, las demencias frontotemporales llevan a que haya más 4R y en Alzheimer se observa una mayor proporción de 3R o de 4R”, agrega.
Pero, ¿por qué se produce este desbalance en las isoformas? “Tau es una proteína con niveles de expresión muy regulados, y se sabe que las causas más frecuentes de los desbalances de las formas 3R y 4R son mutaciones genéticas que influyen sobre estos mecanismos de regulación y hacen que haya más de una que de otra”, cuenta Elena Avale, investigadora adjunta del Consejo en el Instituto de Investigaciones en Ingeniería Genética y Biología Molecular “Dr. Héctor N. Torres” (INGEBI) y también coordinadora del trabajo.
“Durante la investigación encontramos mecanismos afectados por el desbalance entre las formas de Tau, que pueden explicar por qué frente a ciertas mutaciones genéticas se desencadena el proceso neurodegenerativo. Este conocimiento, producto de la investigación básica, podría contribuir a desarrollar una terapéutica a futuro”, agrega.
El gen que codifica para Tau está en el cromosoma 17 y se descubrió que puede dar origen a diferentes formas de la proteína, entre ellas la 3R y 4R. La R significa los puntos de unión a los microtúbulos: 3R tiene 3, y por lo tanto se une y separa de los microtúbulos más fácilmente, mientras que 4R tiene 4 y la adhesión es más estable. Las formas 3R y 4R se producen por un proceso llamado splicing alternativo, por lo cual una parte del ARN mensajero que codifica para uno de los dominios de unión a microtúbulos, se elimina de manera diferencial según el momento del desarrollo o el estado neuronal.
Los investigadores encontraron que 3R favorece el transporte desde el cuerpo hacia el axón, mientras que 4R lo hace desde la sinapsis hacia el cuerpo.
“Durante el desarrollo prevalece 3R, y es lógico si se analiza que una neurona que está creciendo y busca insertarse en el medio tiene que llevar más cosas que las que trae para poder formar las conexiones neuronales. Sin embargo, en el cerebro adulto, con esas conexiones ya formadas y funcionando es necesario generar más estabilidad de los microtúbulos y empezar a traer moléculas desde el axón al soma. Esto se relaciona con la función de 4R que encontramos, que favorece el transporte desde las sinapsis hacia el cuerpo”, explica Avale.
Durante los estudios trabajaron con neuronas humanas diferenciadas a partir de células madre, a las cuales pudieron modularles los niveles de 3R y 4R mediante herramientas de ingeniería genética, y encontraron que cuando en la célula madura se modificaba esa relación 50-50 entre 3R y 4R aparecen defectos en el transporte de vesículas sobre los microtúbulos. Estos defectos podrían estar directamente relacionados con el inicio de procesos neurodegenerativas en algunos tipos de demencias o parkinsonismo.
“Algunas mutaciones en el ADN de la célula llevan a que se modifiquen las proporciones de las formas de Tau, con lo cual si con este sistema se puede regular la síntesis de la proteína para que se mantenga la proporción 50-50 se puede pensar en una plétora de patologías que se podrían corregir”, dice Falzone -quien además colabora como investigador en el Instituto de Bióloga y Medicina Experimental (IBYME, CONICET) y es docente en la Cátedra de Histología de la Facultad de Medicina (UBA)- y agrega: “Este trabajo aporta claves para entender las funciones de 3R y 4R durante el desarrollo, su balance y su rol en las patologías y, a futuro, poder usar esa información para desarrollar tratamientos”.
Pero Tambien la TAU es un macrófago y en alguna ocasión se comporta de manera plural
Se la considera un macrófago y como tal tiene la misión de englobar partículas y destruirlas. Estas particulas algunos autores amantes de la teoría infecciosa de la enfermedad de Alzheimer, creen que son gérmenes y como en otras enfermedades neurodegenerativas, al depositarse sobre estos gérmenes, también lo hacen sobre estructuras cerebrales y bloquean sus funciones.
De forma que el germen o los gérmenes y la proteína TAU depositadas sobre ellos, lesionarían funciones cerebrales fundamentales e inducirían a la demencia, de la misma forma que la B amiloidotica se precipita extracelularmente y mutila también áreas del cerebro
Un equipo de investigadores han descubierto que TAU, una proteína involucrada en la enfermedad neurodegenerativa, tiene un efecto protector capaz de frenar el desarrollo del cáncer cerebral, o glioma.
La capacidad de los tumores para general vasos sanguíneos es fundamental y también lo es en los GLIOMAS para crecer y hacerlos mas agresivos.
En este trabajo los investigadores han demostrado que la proteína TAU está presente en los gliomas menos agresivos. Cuando la cantidad de esta proteína disminuye, los tumores tienden a desarrollar más vasos sanguíneos y volverse más agresivos.
Jesús Ávila, investigador del Centro de Biología Molecular Severo Ochoa (CBMSO-CSIC) y coautor del estudio afirma que “Esta proteína se expresa en las células de glía, mientras que en el ALZHÉIMER solo la vemos expresada en las neuronas,”,. “En los gliomas la proteína tau tiene un factor protector, a más proteína, menos proliferación tumoral”,.
El equipo ha usado muestras de cáncer cerebral de 180 pacientes de los hospitales 12 de Octubre, Gregorio Marañón y Ramón y Cajal, en Madrid, y la Fe, en Valencia. También ha corroborado el hallazgo con muestras de otros 700 pacientes de una cohorte recopilada por la Universidad de California. En un experimento con células de pacientes con glioma y con ratones que sufren esos tumores, los investigadores han descubierto que un fármaco derivado del taxol ya estudiado para otros usos oncológicos es capaz de imitar el papel protector de la tau, frenar el avance de los gliomas y hacer los tumores más vulnerables a la quimioterapia.
Pero que tienen que ver la supuesta condición de macrófago y su deposito intracelular, con la inhibición en los gliomas de la neoangiogenesis.
“Los tumores cerebrales agresivos son muy desalentadores”, este estudio abre una nueva vía por la que buscar un nuevo tratamiento, aunque aún harán falta años de investigación.
A largo plazo El taxol es un compuesto poco tóxico y podría tener un impacto sumado a los tratamientos convencionales”, pero antes habría que demostrar que el fármaco funciona en varios ensayos clínicos con pacientes,
Más a corto plazo, el estudio aporta un conocimiento básico importante. Los investigadores han dilucidado que la proteína Tau ejerce su efecto beneficioso porque refuerza los microtúbulos, que son parte del esqueleto de las células. Una de las primeras cosas que hace el cáncer para proliferar es debilitar ese esqueleto, lo que a su vez facilita que una célula pueda empezar a generar hijas por un proceso de mitosis: La expresión de Tau impide que esa maquinaria de proliferación se ponga en marcha.
“Creemos que tau es una llave entre ALZHÉIMER Y CÁNCER”, resume Gargini, biólogo molecular de origen argentino. Uno de los objetivos ahora es entender si esa llave funciona también a la inversa y puede ofrecer alguna esperanza en esta enfermedad neurodegenerativa que sufren unos 50 millones de enfermos en todo el mundo y de la que se espera que su incidencia se triplique para 2050, pues está asociada al envejecimiento de la población.
Aunque el alzhéimer se descubrió hace más de un siglo, se ignoran sus causas y hoy por hoy no existe ningún tratamiento efectivo. Grandes compañías farmacéuticas han invertido sumas millonarias en intentar desarrollar nuevos fármacos. Todas han fracasado, sobre todo porque su principal táctica, acabar con las proteínas amiloides que se acumulan sin freno en el cerebro de los pacientes, no ha resultado efectiva.
Tras este fracaso ha llegado el momento de la proteína Tau, que tiene una variante normal que no causa ningún problema. Pero en el cerebro de pacientes con alzhéimer la tau está mal plegada, con lo que no puede ser eliminada y pasa a acumularse en ovillos hasta que acaba matando a las neuronas. «Uno de los indicadores precoces del alzhéimer es que los vasos sanguíneos que protegen al cerebro se vuelven permeables. Ahora vemos que tau tiene una función que limita el desarrollo de vasos sanguíneos aberrantes en cáncer. Esto abre nuevas preguntas sobre su papel en el alzhéimer. Nuestra opinión es que la proteína tau funciona tal vez como un guardián del cerebro en su forma normal»,.
Bibliografia
Ricardo Gargini, y Pilar Sánchez-Gómez, investigadores del Instituto de Salud Carlos III de Madrid.ISCIII
The Journal of Neuroscience , liderado por investigadores de Argentina, en colaboración con científicos de la República Checa y del Reino Unido, muestra que sutiles alteraciones en los niveles relativos de Tau afectan de manera significativa el transporte de proteínas relacionadas con la enfermedad de Alzheimer.
Nuño Domínguez es cofundador de Materia, la sección de Ciencia de EL PAÍS. Es licenciado en Periodismo por la Universidad Complutense de Madrid y Máster en Periodismo Científico por la Universidad de Boston (EE UU). Antes de EL PAÍS trabajó en medios como Público, El Mundo, La Voz de Galicia o la Agencia Efe.