ADN TUMORAL. SU INHIBICION
Imagen: Células tumorales. Izzat Suffian, Pedro Costa, Stephen Pollard, David McCarthy & Khuloud T. Al-Jamal.
En condiciones basales todas las celulas tienen mecanismo de reparacion de su ADN, que se altera frecuentemente a lo largo de su vida. Este mecanismo lo tienen tambien las celulas tumorales, pero al multiplicarse muy rapidamente, estos mecanismo reparadores son muy debiles.
La radioterapia y quimioterapia en general, destruyen estos mecanismo celulares tumorales, e impiden su reparacion.
El tratamiento químico del cáncer comenzó con la comprensión de que los agentes dañinos del ADN, como el gas mostaza, presentan propiedades antitumorales notables
Un ensayo clínico de un compuesto que impide la proliferación celular en células cancerosas ha mostrado resultados prometedores en pacientes con tumores sólidos avanzados. Los resultados de este ensayo clínico fueron publicados en la revista Cancer Discovery.
La inhibición de ATR reduce la proliferación de diferentes tipos de cáncer con deficiencias en la reparación del ADN
Una amplia gama de investigaciones preclínicas y una extensa literatura de base científica respaldan el desarrollo de los inhibidores de la quinasa ATM y ATR
Las técnicas de secuenciación actuales pueden identificar mutaciones en genes DDR que pueden afectar la respuesta de las células cancerosas a los inhibidores de ATM o ATR.
Sin embargo, sin estudios funcionales, las consecuencias de estas mutaciones son difíciles de predecir para proteínas como, por ejemplo, ATM, MRE11, RAD50 o FANCD. Adicionalmente, Los defectos de DDR pueden surgir por mecanismos epigenéticos y / o postranscripcionales. Por lo tanto, será esencial desarrollar biomarcadores que puedan determinar de manera sólida el estado funcional de las vías DDR en los tumores y ayudar a diferenciar entre mutaciones nocivas y benignas.
Para los estudios de combinación, es probable que un tema clave sea la preocupación por el potencial de aumento de la toxicidad del tejido normal, y la identificación de las combinaciones y programas que tienen el mayor potencial de efectos selectivos de tumores es un área prioritaria de investigación que necesita más investigación. Los inhibidores selectivos de ATR se encuentran actualmente en fase de desarrollo clínico 1 y parece probable que los inhibidores de ATM con buenas propiedades farmacológicas estén disponibles en un futuro próximo. En particular, los inhibidores de otros objetivos de DDR, incluidos PARP, CHK1 / 2, WEE1 y DNA-PKcs, también se encuentran en desarrollo clínico y será de gran interés seguir el progreso de estos enfoques y utilizar los resultados emergentes para guiar mejor el futuro. desarrollo clínico de inhibidores de ATM y ATR.
En consecuencia, el desarrollo temprano de fármacos se centró en productos químicos genotóxicos, algunos de los cuales todavía se utilizan ampliamente en la clínica. Sin embargo, la eficacia de tales terapias a menudo está limitada por los efectos secundarios de estos medicamentos en las células sanas. Un refinamiento a este enfoque es utilizar compuestos que pueden explorar la presencia de daño de ADN en las células cancerosas. Dado que el estrés por replicación (RS) es una fuente importante de inestabilidad genómica en el cáncer, la focalización de la ataxia quinasa de respuesta RS telangiectasia y la proteína relacionada con Rad3 (ATR) ha surgido como una alternativa prometedora. Con los inhibidores de ATR que ahora entran en ensayos clínicos, aquí revisamos la biología detrás de esta estrategia y discutimos posibles biomarcadores que podrían ser utilizados para una mejor selección de pacientes que responden a la terapia.
En el estudio, un equipo de investigadores del Instituto de Investigación del Cáncer de Londres y la Fundación Royal Marsden NHS ha probado, por primera vez, un potencial tratamiento para frenar la proliferación celular en tumores sólidos avanzados con mutaciones en el gen ATM.
La función de ATR es esencial para la supervivencia de tumores con mutaciones en ATM
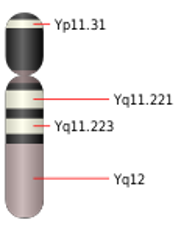
El gen ATM, ubicado en el cromosoma 11 humano, está relacionado con el control de la división celular y la reparación del ADN. Se han relacionado diferentes mutaciones en este gen con el Síndrome de Louis-Barr, así como con un mayor riesgo de padecer cáncer de estómago, vejiga y páncreas entre otros.
En células tumorales con defectos en la reparación del ADN, la actividad de otra enzima, codificada por el gen ATR, es esencial para su supervivencia. Es el caso de los tumores con mutaciones en ATM, en los que la actividad de la enzima ATR compensa sus defectos en la reparación del ADN. Por tanto, inhibir la actividad de ATR resulta una potencial estrategia terapéutica contra este tipo de cánceres.
En estudios anteriores, se había comprobado que la función del gen ATR es esencial en el desarrollo embrionario, puesto que la inhibición total del gen resulta fatal en modelos embrionarios de ratón. No obstante, inhibir la actividad de ATR en ratones adultos modelo no solo es tolerable, sino que reduce considerablemente la proliferación de diferentes tipos de cánceres con deficiencias en los sistemas de reparación del ADN.
Un nuevo fármaco inhibidor de ATR
El tratamiento utilizado en este estudio está basado en la acción de BAY 1895344, una molécula inhibidora de la enzima ATR, relacionada con la reparación del ADN en tumores con defectos en genes como ATM. La actividad antitumoral de este compuesto ya fue probada anteriormente en estudios con modelos animales para diferentes tipos de cáncer.
El Dr. Johann de Bono, profesor en el Instituto de Investigación del Cáncer en Londres, evaluo la seguridad y potencial terapéutico de BAY 1895344 en 21 pacientes con diferentes tipos de tumores sólidos avanzados. Todos los pacientes presentaban una o más alteraciones en el gen ATM y/o en la función de la enzima que codifica y los resltados mostraron una disminución en la proliferación celular de los tumores de 8 de los 21 pacientes. Además, el tratamiento redujo los tumores de 4 de los 13 pacientes restantes.
El principal efecto secundario observado en el estudio fue anemia, aunque en algunos casos los pacientes experimentaron neutropenia (niveles bajos de glóbulos blancos en sangre), trombocitopenia (niveles bajos de plaquetas en sangre), fatiga y náuseas. Muchos de estos efectos secundarios se revirtieron al administrar dosis menores de BAY 1895344.
Los resultados de este ensayo clínico sitúan a BAY 1895344.como un potencial fármaco contra la proliferación de diferentes tipos de cáncer en pacientes con mutaciones en ATM. «Nuestro nuevo ensayo muestra que este nuevo y prometedor tratamiento es seguro y puede beneficiar a algunos pacientes incluso con cánceres muy avanzados”.
La terapia dirigida es un tipo de tratamiento contra el cáncer que usa medicamentos para identificar y atacar a las células cancerosas causando poco daño a las células normales. Estas terapias atacan el funcionamiento interno de las células cancerígenas; la programación que hace que éstas sean diferentes de las células normales y sanas. Cada tipo de terapia dirigida actúa de forma diferente, aunque todas cambian la manera en que una célula cancerosa crece, se divide, se repara por sí misma, o interactúa con otras células.
Inhibidores de PARP
El rucaparib (Rubraca) y olaparib (Lynparza) son medicamentos que se conocen como inhibidores PARP (poli(ADP)-ribosa polimerasa). Las enzimas PARP normalmente están involucradas en un proceso que ayuda a reparar el ADN dañado del interior de las células. Los genes BRCA (BRCA1 y BRCA2) también están normalmente involucrados en otro proceso de reparación de ADN, y las mutaciones de estos genes pueden obstruir este proceso. Al bloquear el proceso de PARP, estos medicamentos dificultan en gran medida que las células del tumor con un gen BRCA anormal reparen el ADN dañado, lo cual a menudo resulta en la muerte de estas células.
El rucaparib (Rubraca) se puede emplear contra el cáncer de próstata en etapa avanzada que sea resistente a castración y que haya crecido a pesar de haber sido tratado con quimioterapia con un taxano (como docetaxel o cabazitaxel) o que no haya respondido a tratamiento con antiandrógenos. Puede utilizarse en hombres que presenten mutación en uno de los genes BRCA. Este medicamento se administra con un agonista de la LHRH o en hombres que se hayan sometido a una orquiectomía.
El olaparib (Lynparza) se puede emplear contra el cáncer de próstata en etapa avanzada que sea resistente a castración y que haya crecido a pesar de haber sido tratado con enzalutamida o abiraterona, medicamentos propios de la terapia hormonal. Puede utilizarse en hombres que presenten mutación en uno de los genes BRCA. Este medicamento se administra con un agonista de la LHRH o en hombres que se hayan sometido a una orquiectomía.
Efectos secundarios de los inhibidores de PARP
Antecedentes
Los fármacos de quimioterapia convencional actúan sobre la división celular al dañar el ADN de las células. Como las células cancerosas se dividen muy rápidamente, estos fármacos afectan a las células cancerosas a un mayor grado que a las células normales. Poder reparar el ADN es vital para la supervivencia de las células y las células normales tienen más de una vía de reparación del ADN. Sin embargo, las células cancerosas a menudo tienen defectos en las vías de reparación que las hace más susceptibles al daño del ADN. Los inhibidores de la PARP son un nuevo tipo de medicación que funciona al impedir que las células cancerosas reparen su ADN una vez que han sido dañadas por la quimioterapia.
Resultados principales
Se buscó en la bibliografía desde 1990 hasta mayo de 2014 y se encontraron cuatro ensayos aleatorios de inhibidores de la PARP versus otros tratamientos o placebo. También se encontraron cuatro estudios en curso. Los cuatro estudios finalizados incluyeron 599 pacientes con cáncer de ovario epitelial recidivante; tres incluyeron pacientes con enfermedad sensible al platino (recidiva de la enfermedad más de 12 meses después del último tratamiento con quimioterapia), y uno incluyó pacientes con enfermedad resistente al platino y parcialmente sensible al platino (recidiva de la enfermedad menos de seis meses o de seis a 12 meses después del último tratamiento con quimioterapia). Los cuatro estudios tuvieron datos que se pudieron agrupar en los análisis. Tres estudios probaron un inhibidor de la PARP conocido como olaparib y un estudio con solamente 75 pacientes probó el veliparib. Como promedio, cuando se agregó al tratamiento convencional, olaparib desaceleró la progresión de la enfermedad en las pacientes con enfermedad sensible al platino en comparación con placebo o ningún tratamiento agregado, pero no afectó la duración de la supervivencia general de las pacientes. Los eventos adversos de cualquier grado fueron frecuentes en el grupo de inhibidor de la PARP y en el grupo control; sin embargo, los eventos adversos graves fueron más frecuentes en el grupo olaparib que en el grupo control cuando se administró como tratamiento de mantenimiento después de un ciclo de quimioterapia. Los eventos adversos graves más frecuentes fueron anemia y fatiga. Veliparib tuvo pocos efectos secundarios graves, pero el número de participantes fueron demasiado pequeño para establecer conclusiones significativas.
La revision de la literatura reciente permite ver como los inhibidores de las enzimas reparadores del ADN tumoral, podrian ser efectivas en el tratamiento de tumores solidos.
Bibliografia
Nature Reviews Volumen de cáncer 18, páginas586–595(2018
. PUBLICADO EL NOVIEMBRE 16, 2020
Rubén Megía González, Genotipia
Yap, T. A. et al. Primer ensayo en humano de la ataxia oral telangiectasia y el inhibidor relacionado con Rad3 BAY 1895344 en pacientes con tumores sólidos avanzados. Discov de cáncer. 2020 Sep 28:CD-20-0868. doi: http://dx.doi.org/10.1158/2159-8290.CD-20-0868
Reparación del ADN de cánceres avanzados. Instituto de Investigación del Cáncer en Londres. https://www.icr.ac.uk/news-archive/new-drug-targeting-dna-repair-shows-promise-in-range-of-advanced-cancers
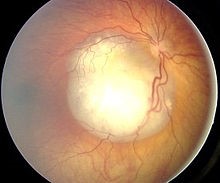 TRATAMIENTO DEL RETINOBLASTOMA PEDIÁTRICO CON UN VIRUS ONCOLÍTICO
TRATAMIENTO DEL RETINOBLASTOMA PEDIÁTRICO CON UN VIRUS ONCOLÍTICO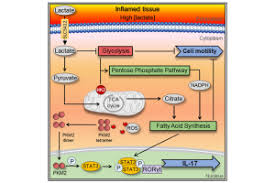 EL LACTATO PUEDE PROVOCAR CÁNCER
EL LACTATO PUEDE PROVOCAR CÁNCER EL ATLAS MÁS COMPLETO DEL GENOMA DEL CÁNCER ADN BASURA’
EL ATLAS MÁS COMPLETO DEL GENOMA DEL CÁNCER ADN BASURA’

 LA PROTEÍNA TAU, , TAMBIÉN SE EXPRESA EN LOS GLIOMAS
LA PROTEÍNA TAU, , TAMBIÉN SE EXPRESA EN LOS GLIOMAS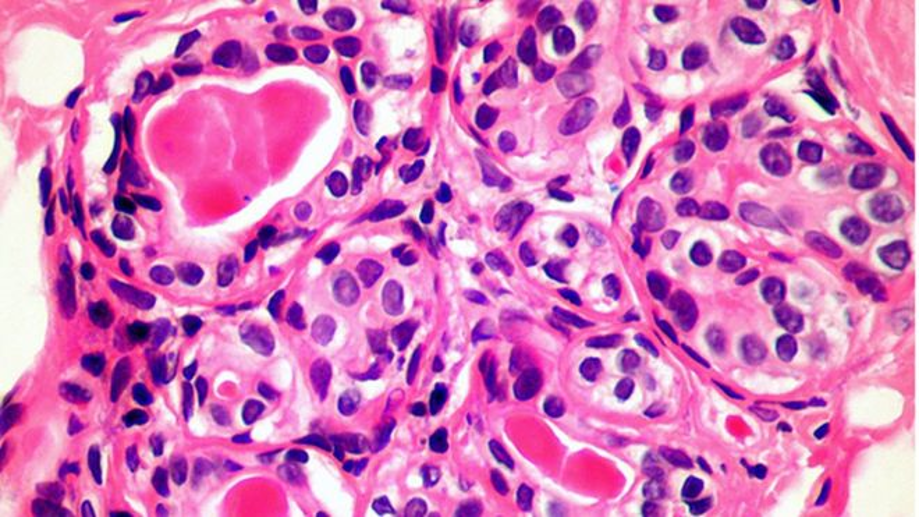
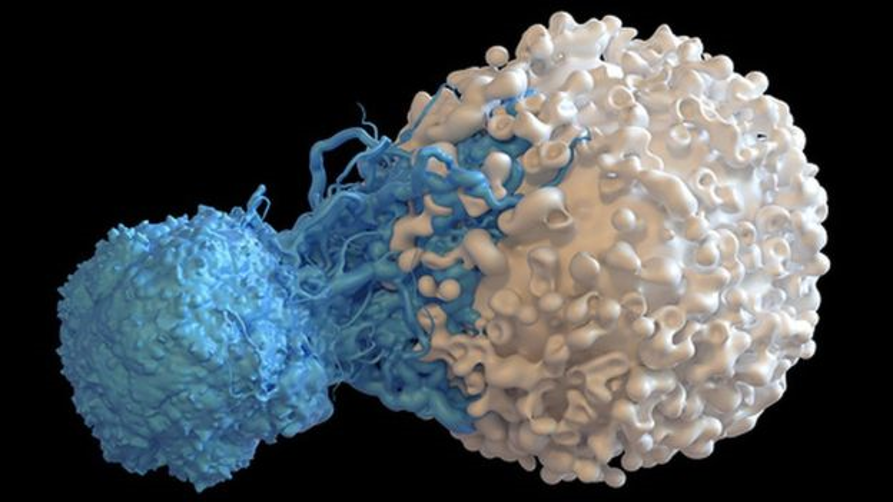 UNA SOLA CÉLULA T PUEDE ENCONTRAR Y MATAR A MÚLTIPLES CÉLULAS CANCEROSAS
UNA SOLA CÉLULA T PUEDE ENCONTRAR Y MATAR A MÚLTIPLES CÉLULAS CANCEROSAS