 TRATAMIENTO DEL RETINOBLASTOMA PEDIÁTRICO CON UN VIRUS ONCOLÍTICO
TRATAMIENTO DEL RETINOBLASTOMA PEDIÁTRICO CON UN VIRUS ONCOLÍTICO
El retinoblastoma es un tumor canceroso que se desarrolla en la retina causado por una mutación en la proteína Rb, codificada por un gen supresor tumoral denominado RB1. Este tumor se presenta en mayor parte en niños pequeños y representa el 3% de los cánceres padecidos por menores de quince años.
Leucocoria del ojo derecho indicando la presencia de tumor.
El retinoblastoma es un tumor canceroso que se desarrolla en la retina causado por una mutación en la proteína Rb, codificada por un gen supresor tumoral denominado RB1.1 Este tumor se presenta en mayor parte en niños pequeños y representa el 3% de los cánceres padecidos por menores de quince años. Constituye la primera causa de malignidad intra-ocular primaria en los niños y la incidencia anual estimada es de aproximadamente 4 por cada millón de niños [1].
El mal puede ser hereditario o no serlo. La forma heredada puede presentarse en uno o ambos ojos y, generalmente, afecta a los niños más pequeños. El retinoblastoma presente sólo en un ojo es no hereditario y afecta prevalentemente a niños mayores. Cuando la enfermedad se presenta en ambos ojos, es siempre hereditaria. Debido al factor hereditario, los pacientes y sus hermanos deben someterse a examen periódicamente, incluyendo terapia genética, para determinar el riesgo que tienen de desarrollar el cáncer.
El retinoblastoma tiende a extenderse hacia el cerebro y la médula ósea, y más raramente se disemina por los pulmones. Estos son factores de pronóstico adverso, así como la invasión coroidal y a través del nervio óptico.1
Leucocoria en un niño con retinoblastoma.
Ojos cruzados en un niño con retinoblastoma.
El retinoblastoma es hereditario en el 40% de los casos; estos niños heredan un alelo mutado (primer evento o hit) en el locus retinoblastoma (RB1) a través de las células germinales. Una mutación somática o cualquier otra alteración en una única célula de la retina da lugar a la pérdida de la función del alelo normal restante, lo que inicia el desarrollo de un tumor. Este trastorno se hereda de manera dominante debido a la presencia de un elevado número de retinoblastos primordiales y su rápida tasa de proliferación, lo cual hace que sea muy probable que se produzca una mutación somática (segundo hit) en uno o más de los retinoblastos existentes. Dado que las posibilidades del segundo evento en la forma hereditaria son tan elevadas, este evento ocurre con frecuencia en más de una célula, de manera que los heterocigotos para esta enfermedad sufren a menudo tumores múltiples que afectan a ambos ojos. Por otro lado, la aparición del segundo evento es un fenómeno de tipo casual y no ocurre en todos los casos; por lo tanto, la penetrancia del gen del retinoblastoma es elevada aunque incompleta. Debido a que la mutación somática del segundo alelo que produce la pérdida de función ocurre con alta frecuencia, las familias que segregan un alelo mutado de un gen supresor de tumores (como RB1) presentan una herencia autosómica dominante de la predisposición al cáncer.2
El 60% restante de los casos es de carácter esporádico (no hereditario), en este caso ambos alelos RB1 de una célula han sido inactivados de forma independiente. En estos casos, generalmente el retinoblastoma sólo se localiza en un ojo. Una diferencia entre los tumores hereditarios y esporádicos es el hecho de que la edad promedio de los pacientes cuando se inicia la forma esporádica pertenece a la primera niñez, es decir, más tarde que la de los lactantes con la forma hereditaria.
Por tanto, existen dos formas de retinoblastoma: una forma bilateral (en la que aparecen tumores independientes en los dos ojos), familiar, y una forma unilateral (en la que aparece un solo tumor en uno de los ojos), esporádica.3 Los afectados con el primer tipo tienen una probabilidad 6 veces mayor de desarrollar otros tipos de cáncer durante su vida, sobre todo osteosarcoma.4 Esto se debe a que en las formas familiares, todos los tejidos somáticos presentan un alelo mutado, por lo que sólo necesitan una segunda mutación para provocar la pérdida de función, mientras que en los casos esporádicos hacen falta dos eventos en cada tejido, lo cual ocurre con menor frecuencia. La pérdida de un alelo se puede producir de diversas maneras:5
pérdida del cromosoma 13 completo;
pérdida del cromosoma 13 normal y duplicación del 13 con el gen mutado;
recombinación homóloga entre los dos cromosomas 13 durante meiosis;
adquisición de una mutación independiente en el segundo alelo de RB1;
En todos los casos, excepto en la mutación independiente, se perderían además todos los marcadores genéticos localizados alrededor de RB1 (y por tanto, ligados a este gen), una situación que se denomina pérdida de heterocigosidad (LOH por sus siglas en inglés, lost of heterozigosity).
Un estudio estadístico realizado por el Dr. Alfred G. Knudson y por el Dr. Thaddeus P. Dryja [2] de los casos de retinoblastoma, para explicar precisamente el mecanismo hereditario de este tumor,65 permitió definir el modelo de funcionamiento de los genes supresores de tumores e identificar el primero de estos genes, RB1. Knudson ganó el Premio Albert Lasker a la investigación médica por este trabajo.
Clínica
Uno de los hallazgos más característicos es la leucocoria; de hecho, es la segunda causa más frecuente de leucocoria en niños tras la catarata congénita. También se presenta estrabismo por afectación del área macular, inflamación del segmento anterior, glaucoma, desprendimiento exudativo de retina y proptosis cuando afecta órbita.
Tratamiento
Imagen histórica mostrando a Gordon Isaacs, el primer paciente tratado con el acelerador linear por retinoblastoma, en 1957. El ojo derecho de Gordon fue resecado el 11 de enero, 1957 ya que el cáncer se había propagado. El ojo izquierdo, en cambio, tenía solamente un tumor localizado que urgió al Dr. Henry Kaplan a intentar tratarlo con el haz de electrones.
La elección de tratamiento que haga el paciente dependerá de cuánto se extienda el mal dentro del ojo y más allá de éste.1El tratamiento de elección es la cirugía, aunque otras opciones de tratamiento con quimioterapia, termo o crioterapia. En un intento de preservar la visión el ojo menos afectado puede tratarse con radioterapia.
En los últimos años se ha ido desarrollando una técnica conocida como quimioterapia intraarterial supraselectiva que se está extendiendo por todo el mundo debido a sus buenos resultados, su disminución de los efectos secundarios y la mejora en la calidad de vida de los pacientes. Dicha técnica consiste en la introducción de un microcatéter desde la femoral hasta la propia arteria oftálmica, lo que permite aplicar una quimioterapia localizada. De este modo se evitan las repercusiones sistémicas de la quimioterapia tradicional, y además se pueden aumentar las dosis del fármaco quimioterápico hasta niveles que serían mortales si se diesen por vía sistémica.78
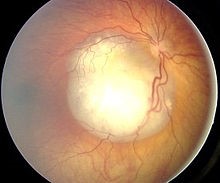
Aspecto funduscópico del retinoblastoma.
Aspecto del retinoblastoma trilateral a la RMN.
El virus ataca y destruye las células cancerígenas del retinoblastoma o cáncer de la retina en modelos animales
Uno de los pacientes afectado por un retinoblastoma con leucocoria, síntoma del tumor ocular.
Un equipo de investigadores del Hospital Sant Joan de Déu Barcelona ha desarrollado un nuevo tratamiento experimental para el retinoblastoma o tumor de la retina, una enfermedad que supone el 11% de los tumores malignos en los niños menores de un año. El tratamiento consiste en inyectar un virus modificado genéticamente dentro del ojo afectado por el tumor. El virus selecciona, ataca y destruye las células cancerígenas, y se aplica en los niños cuyos tumores no responden a los tratamientos convencionales. El trabajo se ha publicado en la portada de la prestigiosa revista Science Translational Medicine y ha merecido uno de los premios más prestigiosos de la oncología pediátrica mundial: el premio Odile Schweisguth de la Sociedad Internacional de Oncología Pediátrica (SIOP).
La investigación ha reproducido experimentalmente tumores obtenidos de pacientes que no se habían curado con los tratamientos actualmente disponibles. Los investigadores de Sant Joan de Déu y de la compañía biotecnológica VCN Biosciences han demostrado que el virus oncolítico VCN-01, desarrollado por modificación genética del adenovirus tipo 5 – un virus común que normalmente causa síntomas de un resfriado – es capaz de infectar y multiplicarse solo en los tumores y no en células sanas de la retina. La selectividad del virus por los tumores se basa en el funcionamiento anormal del gen del retinoblastoma (RB1) en las células afectadas por el tumor, en las que se produce un aumento de la cantidad libre de una molécula denominada E2F-1.
El Hospital Sant Joan de Déu ha iniciado un ensayo clínico, dirigido por los doctores Guillermo Chantada, Jaume Catalá y Jaume Mora, para tratar con el virus oncolítico VCN-01 a pacientes con tumores oculares quimio-resistentes. El objetivo de este estudio experimental, en el que también participa VCN Biosciences, es describir la seguridad del tratamiento y obtener los primeros indicios de su eficacia clínica.
El nuevo tratamiento se aplica a los casos de retinoblastoma más agresivos
El cáncer de retina se diagnostica cada año a 8.000 niños en todo el mundo. Es el tumor ocular más frecuente en la población infantil. En la actualidad, cuando está indicada la preservación ocular, los niños reciben en una primera fase quimioterapia intraarterial, que se aplica a través de un largo y fino catéter introducido por la arteria femoral (en la ingle) y conducido hasta la arteria oftálmica para, una vez allí, administrar localmente la quimioterapia.
En ciertas ocasiones, además, se inyecta directamente quimioterapia dentro del ojo, en el denominado humor vítreo. En un 30% de los casos, sin embargo, el tumor no responde a ninguno de estos dos tratamientos y los oftalmólogos no tienen otra opción que extirpar el ojo afectado para evitar que el cáncer se extienda a otros órganos del cuerpo, ya que entonces las posibilidades de curación son muy bajas. Esta nueva terapia con virus pretende evitar la extirpación ocular y disminuir los casos de ceguera en pacientes con retinoblastoma.
Este nuevo tratamiento forma parte del conjunto de nuevas terapias avanzadas que está poniendo en marcha el Hospital Sant Joan de Déu y que representan un nuevo paradigma que permite la personalización del tratamiento de algunos tipos de cáncer.
El Hospital tiene abiertos varios ensayos clínicos basados en terapias innovadoras dirigidas al tratamiento del glioma difuso del tronco cerebral (un cáncer que hoy por hoy es incurable) el retinoblastoma (el cáncer de retina) y las leucemias linfoblásticas agudas de tipo B, tratadas en algunos casos con CART-19. Esta lína de trabajo se enmarca en el proyecto del futuro SJD Pediatric Cancer Center, concebido como un centro de asistencia e investigación traslacional, orientado a la puesta en marcha de nuevos tratamientos.
Bibliografía
↑ Saltar a:a b c Kumar, MBBS, MD, FRCPath, V.; Abul K. Abbas, MBBS, Nelson Fausto, MD and Jon Aster, MD (2009). «Retina and vitreous». Saunders (Elsevier), ed. Robbins & Cotran Pathologic Basis of Disease (8th edición).
↑ Nussbaum, R.L.; R. R. McInnes, H. F. Wilard. (2007). Thompson & Thompson Genetics in medicine. (7th edición). Saunders. ISBN 9781416030805.
↑ Alberts et al (2004). «Biología molecular de la célula». Barcelona: Omega. ISBN 54-282-1351-8.
↑ Kleinerman RA, Tucker MA, Tarone RE, et al. (abril de 2005). «Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: an extended follow-up». J. Clin. Oncol. 23 (10): 2272-9. PMID 15800318. doi:10.1200/JCO.2005.05.054.
↑ Saltar a:a b Gelehrter, T.; et al. (1998). Principles of Medical Genetics (2nd edición). Baltimore: Williams et Wilkins.
↑ Knudson AG (abril de 1971). «Mutation and cancer: statistical study of retinoblastoma». Proc. Natl. Acad. Sci. U.S.A. 68 (4): 820-3. PMC 389051. doi:10.1073/pnas.68.4.820.
↑ «The Evolution of Treatments for Retinoblastoma». Retina Today. Consultado el 29 de noviembre de 2011.
↑ «Superselective ophthalmic artery chemotherapy as primary treatment for retinoblastoma (chemosurgery». Ophtalmology. Consultado el 29 de noviembre de 2011.
ACTIVIDAD CLÍNICA DESTACADAS INVESTIGACIÓN PRENSA
24 ENERO 2019