Se transmite el SARS-CoV-2 por los alimentos congelados?
La predilección del coronavirus por las bajas temperaturas sustenta los temores sobre su pervivencia en carnes y pescados congelados para su distribución.
Los estudios publicados no coinciden sobre la viabilidad infectiva del SARS-CoV-2 adherido a alimentos congelados.
José R. Zárate
La semana pasada, las instalaciones de la cadena de frío en la ciudad portuaria septentrional de Tianjin en China fueron cerradas cuando la infección por SARS-CoV-2 de un trabajador de alimentos congelados de 38 años se vinculó a un cargamento de 28 toneladas de codillos de cerdo procedentes de Alemania. En Wuhan, las autoridades dijeron que habían detectado virus en el embalaje de un lote de carne de vacuno congelada y deshuesada de Brasil. Las aduanas también suspendieron las importaciones de sepia de la compañía india Basu. El mes anterior dos estibadores de Qingdao parece que se contagiaron al manipular bacalao congelado.
Acciones similares se vienen repitiendo desde que China eliminó casi completamente el coronavirus. En la actualidad ha interrumpido importaciones de un centenar de suministradores de 20 países, en especial carne de Brasil y Argentina, sus mayores proveedores.
Las estrictas directrices de China sobre la cadena de frío exigen la «eliminación completa» y la «denegación estricta de la entrada» de cualquier producto sospechoso de contacto con el virus. Obligan a una desinfección meticulosa, incluidos los envases interiores y exteriores, y análisis a todas las mercancías importadas. Incluso prohíben compras en países gravemente afectados por la pandemia. «Si un producto está contaminado, devuelven la totalidad de los alimentos. Es su derecho, pero no creo que sea muy necesario. Bastaría con un proceso de descontaminación», decía a la agencia Reuters Jin Dong-Yan, profesor de virología de la Universidad de Hong Kong.
Quizá tenga algo que ver con esta obsesión el que el primer brote del SARS-CoV-2 se hallara en el mercado de mariscos de Wuhan y que el segundo gran brote, en junio, con 335 casos, se asociara al mercado de mariscos Xinfadi en Beijing (Pekín): el salmón importado desapareció de las tiendas y restaurantes después de que los medios de comunicación informaran de que se había detectado el virus en piezas a la venta en ese lugar y hasta en las tablas de cortar el salmón.
Como aval científico, un equipo de la Universidad de Guangzhou encabezado por Manman Dai publica este mes en Journal of Infectious Diseases un análisis sobre los virus SARS-CoV-2 adheridos al salmón almacenado a 4 ºC, la temperatura a la que se conserva el pescado en cámaras frigoríficas, y también en otras muestras conservadas a 25 ºC. El SARS-CoV-2 unido al salmón permaneció viable durante 8 y 2 días, respectivamente. El almacenado a 25 ºC dio como resultado una infectividad atenuada.
Estas observaciones son consistentes con otros estudios de permanencia del virus en aerosoles o en distintas superficies, como el publicado por el grupo de Van Doremalen, de los Institutos de Salud (NIH) estadounidenses, en abril en el New England: a 21-23 ºC no se encontró SARS-CoV-2 viable después de 4 horas en superficies de cobre, 24 horas en cartón y después de 3 días en superficies de acero inoxidable y plástico, si bien se ha ido observando la pérdida de viabilidad del SARS-CoV-2 con el aumento de la temperatura.
Pero hasta qué punto esos virus posados en superficies inertes son contagiosos sigue analizándose y debatiéndose. Otro estudio prepublicado este mes en MedRxiv por un equipo de la Universidad de Tufts, en Estados Unidos, analizó hisopos tomados de superficies de alto contacto en una ciudad de Massachusetts de abril a junio. De 348 muestras, 29 (8,3 %) fueron positivas para SARS-CoV-2, incluidos los botones de semáforos para el paso de peatones, las asas de los cubos de basura y los picaportes de puertas de entrada a tiendas de comestibles, de licores, de un banco y de una gasolinera. “El riesgo estimado de infección por tocar una superficie contaminada -concluían- era bajo (menos de 5 de cada 10.000), lo que sugiere que estos fómites desempeñan un papel mínimo en la transmisión comunitaria SARS-CoV-2”.
Parecidos resultados obtuvo el equipo de Christian Gortázar, de la Universidad de Castilla-La Macha, en un estudio similar efectuado en Horcajos de los Montes (Ciudad Real): 7 (12,28%) de las 57 muestras y 6 (26%) de los lugares analizados dieron positivo para el ARN del SARS-CoV-2. “El SARS-CoV-2 puede permanecer estable en un entorno favorable”, escribían en su informe en Transboundary and Emerging Diseases. “Sin embargo, estudios de campo recientes que informan sobre la detección de ARN del SARS-CoV-2 a partir de muestras ambientales y el intento de aislamiento del virus no han logrado inducir un efecto citopático, o encontraron solo señales débiles para una replicación competente del virus”.
Y dos estudios publicados por el equipo italiano de Marta Colaneri, de la Universidad de Pavía y el Policlínico San Mateo, uno en marzo en Journal of Hospital Infection y otro en agosto en Clinical Microbiology and Infection, con muestras tomadas en diversas superficies del hospital italiano concluían igualmente en la escasez de virus viables, siempre y cuando se sigan los procedimientos normales de desinfección.
En el segundo estudio, de 26 muestras recogidas solo dos fueron positivas para el ARN del SARS-CoV-2, ambas recolectadas en la superficie de máscaras de presión positiva continua para las vías respiratorias; ninguna indujo un efecto citopático el día 7 de cultivo. Explicaban que, “aunque el contacto diario con superficies inanimadas y fómites de pacientes en áreas contaminadas puede ser un medio de infección, nuestros datos obtenidos en condiciones de la vida real sugieren que podría ser menos extenso de lo que se reconocía hasta ahora”.
Aun así, en China no se fían. Como el pescado importado y exportado debe transportarse a baja temperatura (de 0 a 4 ºC), si está contaminado con SARS-CoV-2 puede servir como fuente de transmisión, dicen los autores de Guangzhou. A diferencia de muchos otros productos alimenticios, el pescado debe transportarse, almacenarse y venderse en un ambiente de baja temperatura. “Esto significa que el virus adherido puede sobrevivir durante mucho tiempo… e infectar los enterocitos intestinales humanos”.
La hipótesis de los alimentos y sus envases como fuente de contagio la analizaba y exponía en agosto un equipo de la Universidad Nacional de Singapur dirigido por Dale Fisher en una prepublicación en BioRxiv. El brote de junio de Beijing ocurrió 55 días después del último caso registrado localmente y, asimismo, Vietnam y Nueva Zelanda tuvieron nuevos brotes inexplicables a los 99 y 102 días, respectivamente, desde sus últimas transmisiones locales identificadas.
Es posible, dicen, que en estas regiones la erradicación nunca se lograra realmente o que se hubiera llevado a cabo una transmisión no identificada a través de algún viajero. “Otra posibilidad es el transporte de productos contaminados como los alimenticios”. Aluden en este sentido a los numerosos brotes aparecidos en instalaciones de procesamiento de carnes y mariscos en Portugal, Alemania, Reino Unido, Ghana o Australia, por citar casos documentados.
El equipo de Singapur escogió varias muestras de salmón, pollo y cerdo, las contaminó con virus y las almacenó a tres temperaturas diferentes: 4 ºC,-20 ºC y -80 ºC. Las fueron analizando después de 1, 2, 5, 7, 14 y 21 días. La cantidad de SARS-CoV-2 se mantuvo constante en las tres temperaturas durante la duración del experimento. Y la infectividad perduró durante tres semanas tanto en las muestras refrigeradas (4 ºC) como en las congeladas (-20 ºC y -80 ºC).
La Organización Mundial de la Salud indica que es muy improbable contraer la covid-19 a partir de alimentos o envases de alimentos, pero, aunque “no sea una vía de infección importante, la posibilidad de que lleguen productos contaminados a una región sin covid-19 e iniciar un brote es una hipótesis importante”, insiste el equipo de Singapur. Añaden que las condiciones de trabajo en mataderos y envasadoras facilitan los contagios.
“Con una carga significativa de virus presente en trabajadores infectados y en el ambiente laboral, la contaminación de la carne con SARS-CoV-2 sería posible durante el procesamiento. Las líneas de matanza generalmente funcionan a temperatura ambiente, pero el proceso más tarde baja a no más de 12 ºC y la carne se mantiene a 3-7 ºC… Nuestro trabajo de laboratorio ha demostrado que el SARS-CoV-2 puede sobrevivir al tiempo y las temperaturas asociados con las condiciones de transporte y almacenamiento habituales en el comercio internacional de alimentos”. Ante esa eventualidad, aconsejaban extremar las medidas de higiene y control tanto en esos lugares de procesamiento como en los mercados receptores en el otro extremo de la cadena de suministro.
Fuera de China, los alimentos congelados rara vez se someten a rastreos de virus. En agosto, un trabajador neozelandés de almacenamiento en frío dio positivo, pero más tarde las autoridades sanitarias descartaron los alimentos congelados como fuente. Algunos científicos han señalado que las pruebas en alimentos de cadena fría y envases también detectan fragmentos muertos del virus, lo que significa que los resultados positivos no indican que la enfermedad sea viable y pueda infectar.
«La gente no debe temer la alimentación, el envasado de alimentos o la entrega de alimentos», dijo en agosto Mike Ryan, jefe del programa de emergencias de la OMS. «No hay evidencia de que la cadena alimentaria esté contribuyendo a la transmisión de este virus». De todos modos, si algo caracteriza a este coronavirus es la manía que tiene a las evidencias. Para los más temerosos, la higiene obligada con los alimentos y su preparación y una adecuada cocción bastarían para eliminarlo.
Categoría: MICROSBIOS (Página 3 de 5)
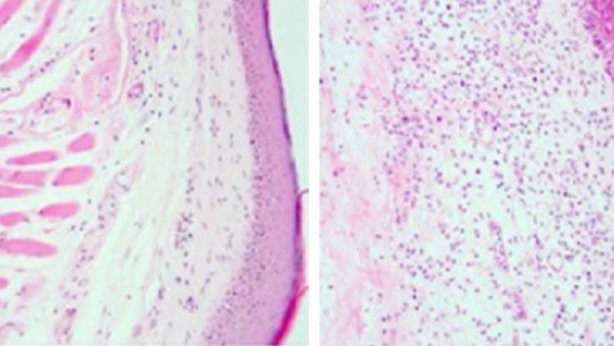 INMUNNIDAD EN LOS VIRUS
INMUNNIDAD EN LOS VIRUS
Cortes histológicos que muestran que en presencia de la proteína viral (izq.) se inhibe una infiltración de células inmunes (puntos oscuros) y mayor inflamación que en ausencia de ella (dcha.). (CSIC)
«Los virus conocen cómo funciona el sistema inmune. «, La magia de la biología, nos lleva a pensar que los virus conocen la inmunidad y están facultados para luchar contra ella.
La revista Nature Communications, publica un trabajo realizado en ratones, han utilizardo el modelo de la viruela, que causó la muerte de millones de personas antes de su erradicación y es la primera enfermedad infecciosa erradicada mediante un programa global de vacunación en 1980.
Los autores descubren que los poxvirus utilizan una estrategia única. Producen una copia de los receptores celulares del factor de necrosis tumoral (TFN) para inhibir la respuesta inmune»,
El TFN está implicado en el inicio y la coordinación de la respuesta inflamatoria y tras unirse a receptores específicos activa células inmunes necesarias para la defensa ante las infecciones. Cuando esta molécula se produce de forma incontrolada y causa una activación crónica de la respuesta inflamatoria, da lugar a enfermedades autoinmunes.
Se sabia que se pueden utilizar versiones solubles de los receptores de TFN en la clínica para tratar enfermedades autoinmunes como la artritis reumatoide.
Los virus también bloquean la respuesta inmune y para ello optimización de los receptores celulares del TFN añadiendo un nuevo dominio, denominado Secret. Este nuevo dominio interacciona con otras moléculas inmunes conocidas como quimiocinas, que controlan la migración de las células inmunes a los sitios de infección e inflamación. Al bloquear las quimioquinas, los virus consiguen que las células inmunes no se dirijan a los tejidos infectados y de esta forma inhiben una respuesta inflamatoria.
El mecanismo combinado contra quimiocinas y el factor de necrosis tumoral, ideado por estos virus, hace que al eliminar la proteína viral pierda la batalla contra el sistema inmune y el virus sea incapaz de causar la enfermedad. De esta forma, el efecto antiinflamatorio de los receptores del TFN se ve potenciado.
La estrategia viral se podría trasladar al campo de la medicina clínica añadiendo el dominio Secret a los receptores del TFN que se utilizan actualmente como medicamentos para frenar una respuesta inmune excesiva y tratar enfermedades autoinmunes. «Es interesante comprobar cómo el conocimiento básico de las estrategias ‘secretas’ utilizadas por el virus de la viruela para evadir nuestras defensas podría utilizarse ahora para mejorar medicamentos que pueden curar enfermedades y mejorar nuestra calidad de vida».
También se han detectado puntos de control en células inmunes que regulan su respuesta a virus, según trabajo del
El CIC bioGUNE, con la Universidad de Vermont, ha identificado puntos de control en la células CD8 que regulan su capacidad de respuesta a agentes infecciosos, en el virus de la gripe.
La proteína mitocondrial MCJ regula el metabolismo de las células CD8 durante las diferentes fases de su respuesta cuando se enfrentan a infecciones: tanto la activación, como la generación de memoria, una vez que el agente infeccioso ha desaparecido. Cuando no existe esa proteína, aparecen muchas más células de memoria inmunitaria.
Este trabajo tiene también la posibilidad de regular el desarrollo de este tipo de células durante la vacunación. Si se pudiera controlar la cantidad de MCJ en las células se podría, por tanto, mejorar teóricamente la eficacia de las células y, con ello, la eficiencia de vacunas que las activan.
El trabajo ha sido dirigido por el grupo de Mercedes Rincón, profesora en el Departamento de Medicina de la Universidad de Vermont, junto con el equipo de Juan Anguita, en el CIC bioGUNE, y se publica en la revista Immunity.
que pertenecen al sistema inmune adaptado y son especialmente aptas para la lucha frente a las infecciones por virus.
El metabolismo y su regulación son claves en el control de la actividad celular. «El objetivo es desarrollar herramientas para el control de la actividad de MCJ que permita acelerar o frenar el metabolismo celular, dependiendo de las necesidades específicas de la patología a la que se quiera hacer frente y que puede implicar células inmunes u otros tipos celulares involucrados en cánceres o enfermedades infecciosas», señala Juan Anguita, investigador Ikerbasque y director del estudio en CIC bioGUNE.
Rincón y Anguita, desde 2007, estudia en MCJ la respuesta de macrófagos -células del sistema inmune innato que se encargan del reconocimiento y destrucción de agentes infecciosos- y células T.
En los dos comentarios que hago sobre los trabajos publicados, en el primero se intente regular la auto imunidad y del segundo incrementado la respuesta de los macrófagos. Esta dualidad forma parte de nuestro tiempo y podría abreviarse diciendo; La inmunidad es un producto delicado que surge de la vitud, ni mucho ni poco, lo suficiente
Bibliografia
ESTUDIO DEL CSIC EN ‘NATURE COMMUNICATIONS’
Madrid | 03/05/2018 19:15
Juan Anguita, investigador del CIC BioGUNE y uno de los autores del trabajo. Antonio Alcamí del CSIC V en el Centro de Biología Molecular Severo Ochoa (centro mixto del CSIC y la Universidad Autónoma de Madrid).
 GÉRMENES Y ARTERIOSCLEROSIS
GÉRMENES Y ARTERIOSCLEROSIS
La existencia de gérmenes en las placas de ateroma es un ideal que al igual que se esta haciendo con la microbiota y las enfermedades degenerativas nos lleva al sueño de fabricar un antigermen, o algo parecido que nos lleve a la eliminación de esta enfermedad, la arterioesclerosis, a la que no podemos vencer por otro camino________________________________________
La arteriosclerosis se ha convertido en la primera causa de mortalidad en Occidente. Así la cifra de defunciones secundarias a enfermedades directamente relacionadas con la arteriosclerosis representan el 30% de la mortalidad total en el mundo, siendo responsables del 25% de las muertes en países en vías de desarrollo y del 50% en los países industrializados(1).
La importante repercusión social y económica del proceso ha hecho que sea este uno de los campos de la medicina donde más se ha trabajado en las últimas décadas.
Fruto de este estudio se ha llegado a la evidencia de que arteriosclerosis es un proceso inflamatorio (2). De hecho las lesiones ateromatosas presentan una serie de cambios específicos secundarios a respuestas tanto celulares como moleculares. La estría grasa, que es el tipo de lesión más temprana en la arteriosclerosis está constituida por macrófagos y linfocitos T. Entre los factores que promueven la arteriosclerosis, y por tanto la placa de ateroma se encuentran la hipercolesterolemia, las LDL oxidadas, el déficit de homocisteína, la hipertensión y determinadas infecciones (3,4).
En esta revisión analizamos los hallazgos que sustentan la relación entre la arteriosclerosis y la bacteria Chlamydia pneumoniae, la controversia que rodea a esta relación así como el estado actual de conocimientos y las perspectivas futuras
La hipótesis de que factores infecciosos estuvieran en relación con la arteriosclerosis viene dada por diferentes razones. La observación de las curvas de incidencia de enfermedad coronaria en EEUU entre 1940 y 1970 simulaba un patrón epidémico similar al de algunas enfermedades infecciosas.
A finales de los años 70 se observó que la infección, de forma experimental, en pollos por un tipo de herpesvirus producía una afectación arterial similar a la arteriosclerosis(5).
La implicación de Helicobacter pylori en el desarrollo del úlcus péptico, una enfermedad tradicionalmente considerada sin relación con la infección contribuyó a aumentar la creencia de que agentes infecciosos estuvieran implicados en otros procesos.
Entre los microorganismos implicados en la progresión de la lesión arteriosclerosa destaca el herpesvirus, Chlamydia y Helicobacter pylori. La mayoría de estudios que relacionan estos microorganismos con la arteriosclerosis se fundamentan en datos seroepidemiológicos basados en la titulación de anticuerpos (6).
Nos centraremos en la relación entre Chlamydia y arteriosclerosis. Chlamydia es una bacteria Gram negativa. En el humano son patógenas tres especies: C. trachomatis, C. psittaci y C. pneumoniae (también conocida como agente TWAR). Es precisamente esta última especie la que se ha relacionado con la arteriosclerosis (7).
La relación Chlamydia-arteriosclerosis viene avalada por diferentes evidencias:
EVIDENCIA SEROEPIDEMIOLÓGICA
Saikku y col describieron en 1988 la elevación de anticuerpos anti-Chlamydia determinados mediante microinmunofluorescencia en pacientes con infarto agudo de miocardio y enfermedades coronarias respecto a un grupo control (8).
Posterior a este estudio distintos autores han corroborado los datos de Saikku.
En la mayoría de estudios publicados se ha encontrado una odds ratio dos veces superior para serología positiva a Chlamydia en pacientes ateroscleróticos. Sin embargo estos estudios utilizan diferentes poblaciones, emplean distintos criterios para la clasificación de casos y controles en cada uno de los estudios y además, no valoran de igual manera las diferentes variables de confusión. La mayoría de estudios publicados a este respecto son descriptivos, por lo que permiten establecer la hipótesis pero no la relación causal. Además los estudios descriptivos no distinguen, generalmente, determinadas variables de confusión. Así por ejemplo el tabaco, un factor de riesgo para patología coronaria, podría predisponer a la infección por Chlamydia pneumoniae y contribuir ala elevación de los anticuerpos (9).
En este tipo de estudios la técnica empleada para la detección y titulación de anticuerpos es la microinmunofluorescencia, que tiene el grave inconveniente de su poca reproductibilidad (10).
EVIDENCIA HISTOPATOLÓGICA
En 1992 Shor y col detectaron, en autopsias, la presencia de Chlamydia pneumoniae en estrías grasas y placas de ateroma (11).
Posteriormente, y a través de distintas técnicas, como la inmunocitoquímica, reacción en cadena de la polimerasa, microscopía electrónica y por cultivo se ha observado Chlamydia en placas de ateroma procedentes de arterias coronarias, carótidas, aorta abdominal, y placas ateroscleróticas, tanto de pacientes jóvenes como de ancianos (12).
Estudios in vitro han demostrado que C. pneumoniae es capaz de infectar y reproducirse en células musculares lisas, células endoteliales coronarias y macrófagos. En 1996, Ramírez aisló Chlamydia en un cultivo procedente de tejido coronario ateromatoso en un paciente sometido a trasplante cardiaco (13). Otros investigadores han aislado Chlamydia en otros tejidos con ateroma (14).
Existen nuevos datos acerca de la presencia de Chlamydia en tejido arterial no ateromatoso. En muchos de los estudios histopatológicos se define la presencia de Chlamydia a través de la existencia de DNA bacteriano, antígenos o cuerpos elementales. Pero ninguno de estos marcadores indica la presencia de bacterias viables.
Además el DNA de Chlamydia pneumoniae se ha detectado en válvulas aórticas estenosadas, vasos hepáticos y bazo, lo que ha llevado a levantar la hipótesis de que pudiera tratarse de un observador inocente presente en el tejido inflamatorio (9). El hecho de haber aislado la bacteria en diferentes tejidos, levanta la hipótesis de si el organismo se disemina y persiste en múltiples tejidos además del tejido cardiovascular. Estos hallazgos sustentan la hipótesis del observador inocente. A este respecto Jackson y colaboradores al analizar la presencia de Chlamydia en diferentes tejidos (cardiovascular, pulmonar, esplénico, médula ósea, hepático,…) procedente de autopsias, encontraron una presencia bacteriana en tejido cardiovascular mayor con respecto al resto de tejidos, siendo esta diferencia estadísticamente significativa. Este resultado refuerza la hipótesis de que Chlamydia pneumoniae juegue un papel en la patogénesis de la enfermedad aterosclerótica cardiovascular (15).
El mecanismo fisiopatológico que explicaría la participación de C. pneumoniae en el proceso aterogénico sería a través de la infección de los macrófagos, ya que se trata de una bacteria intracelular obligada. Bajo determinadas circunstancias Chlamydia desarrollaría una infección crónica en los macrófagos. Los mecanismos que favorecerían dicha infección no han sido aclarados todavía. La infección por distintos microorganismos produciría la transformación de las células musculares lisas y de las células endoteliales de la pared arterial. Así producirían disfunción endotelial, a través del aumento de sustancias procoagulantes, disminución de la fibrinolisis, aumento de la adhesión leucocitaria y aumento en la producción de citokinas. Junto a esto también se produciría disfunción en las células musculares lisas a través del aumento de su proliferación, disminución de los mecanismos de apoptosis, aumento de la esterificación del colesterol y aumento en la producción de citokinas. Todo ello produciría una alteración y reclutamiento de leucocitos (6).
Diferentes estudios han demostrado la capacidad de diferentes virus para modificar las propiedades procoagulantes y anticoagulantes de la célula endotelial. Sin embargo existen pocos datos que soporten la participación de Chlamydia en la regulación de las células endoteliales. Al igual que otras bacterias Gram negativas posee una endotoxina, la lipopolisacaridasa, si bien sus efectos sobre la coagulación son todavía desconocidos.
Las lipopolisacaridasas bacterianas son activadores clásicos de la producción de citokinas. La mayoría de estos estudios se centran en la endotoxina derivada de E.coli. Pocos datos existen sobre la capacidad de Chlamydia en producir efectos similares. Además los resultados obtenidos con E.coli no tienen porque ser extrapolados en su totalidad a otras especies bacterianas (16).
También ha podido demostrarse la capacidad de algunos virus en la transformación de células musculares lisas (17). Actualmente no existen datos que sustenten la capacidad de C. pneumoniae en la modulación o transformación de las células musculares lisas.
Con todo esto solo nos queda saber si la relación entre Chlamydia y aterosclerosis cumple los postulados propuestos por Koch a finales del siglo XIX. Dichos postulados establecen que para considerar a un microorganismo responsable de una enfermedad deben cumplirse las siguientes condiciones: (18)
1. El germen causante de la enfermedad puede ser aislado del individuo afectado.
2. El agente infeccioso puede ser identificado mediante cultivo, o bien a través de microscopio.
3. Al inocular a un huésped susceptible el microorganismo, este es capaz de producir la enfermedad.
En lo referente al primer punto, aunque las placas de ateroma contienen frecuentemente antígenos de Chlamydia o ácidos nucleicos de este, otros casos carecen de la evidencia, aunque sea de forma indirecta de la presencia de C.pneumoniae. Además a pesar de que la bacteria está presente en la lesión eso no implica que participe en la patogenia.
A lo largo de esta revisión ya se ha comentado la identificación de C. pneumoniae en las placas de ateroma, confirmando así el segundo postulado de Koch.
El tercer postulado no ha podido ser demostrado hasta el momento actual (6).
Si bien Chlamydia no cumple las tres condiciones, numerosos autores has puesto en tela de juicio su validez para considerar a un microorganismo responsable de la enfermedad (19).
Si C. pneumoniae juega un papel en la aterogénesis sería lógico pensar que actuando sobre ella podríamos prevenir la enfermedad derivada de la artrioesclerosis. Bajo esta premisa en 1997 se realizó el primer estudio en este sentido (2,6). Para ello se empleo azitromicina, un macrólido de nueva generación, efectivo en infecciones respiratorias por C. pneumoniae, bien tolerado y con excelente perfil farmacocinético respecto de los macrólidos clásicos (20).
En este trabajo se demostró que aquellos pacientes que después de haber padecido un infarto agudo de miocardio presentan títulos elevados de anticuerpos anti-Chlamydia presentan un riesgo cuatro veces superior de padecer nuevos eventos cardiovasculares que aquellos que presentan títulos más bajos.
Además se observó que en el subgrupo de pacientes con títulos de anticuerpos elevados y sometidos a tratamiento con azitromicina no había diferencias estadísticamente significativas respecto al grupo de pacientes con títulos bajos de anticuerpos.
Una de las hipótesis que explicaría esto sería que el antibiótico actuaría erradicando o suprimiendo la infección, lo que conllevaría la estabilización de la placa de ateroma al frenar, en cierta medida, los efectos inflamatorios y procoagulantes atribuidos a Chlamydia.
Posteriores a este estudio se han diseñado otros que valoran la efectividad del tratamiento antibiótico en la patología aterosclerótica (21,22).
CONCLUSIÓN:
En la última década se ha avanzado de manera espectacular en el conocimiento de la patogénesis de la aterosclerosis. Fruto de este avance es la implicación de agentes infecciosos en el proceso. Con estos antecedentes, y la situación actual del problema, es de esperar que los próximos años sean decisivos para conocer la implicación real de Chlamydia pneumoniae en el proceso y de ello derivarse un tratamiento efectivo.
BIBLIOGRAFÍA.
1. World Heart Organization. World Health Organization warns of growing «crisis of suffering». Human and social costs of chronic diseases will rise unless confroted now, WHO Director-General says. 1997. http://www.who.org./whr/1997/presse.htm
2. Gupta S. Chronic infection in the etiology of atherosclerosis, focus on Chlamydia pneumoniae. Atherosclerosis 1999; 143(1): 1-6.
3. Ross R. Atherosclerosis – An inflammatory disease. N Engl J Med 1999; 340 (2): 115-26.
4. Muhlestein JB. Chronic infection and coronary artery disease. Med Clin North Am 2000; 84(1): 123-48.
5. Fabricant CG, Fabricant J, Litrenta MM, Minick CR. Virus-induced atherosclerosis. J Exp Med 1978; 148: 335-40.
6. Libby P, Egan D, Skarlatos S. Roles of infectius agents in atherosclerosis and restenosis. Circulation 1997; 96: 4095-103.
7. Peeling RW. Chlamydiae as pathogens: new species and new issues. Emerging Infectious Diseases 1996; 2(4): 307-19.
8. Muhlestein JB. The link between Chlamydia pneumoniae and atherosclerosis. Infect Med 1997; 14(5): 380-82.
9. Danesh J, Collins R, Peto R. Chronic infections and coronary heart disease: is there a link?. Lancet 1997; 350: 430-36.
10. Wnag SP, Grayston JT. Microimmunofluorescence serology in Chlamydia trachomatis. In: de la Maza LM, ed. The 1983 International Symposium on Medical Virology, New York: Elsevier Science, 1984:87-118.
11. Shor A, KuoCC, Patton DL. Detection of Chlamydia pneumoniae in coronary arterial fatty streaks and athermatous plaques. S Afr Med J 1992; 82: 158-61.
12. Ong G, Thomas BJ, Mansfield AO. Detection and widespread distribution of Chlamydia pneumoniae in the vascular system and its posible implications. J Clin Path 1996; 49: 102-06.
13. Ramirez JA. Isolation of Chlamydia pneumoniae from the coronary artery of a patient with coronary atherosclerosis. The Chlamydia pneumoniae Atherosclerosis Study Group. Ann Int Med 1996; 125: 979-82.
14. Taylor-Robinson D, Thomas BJ. Chlamydia pneumoniae in atherosclerotic tissue. J Infect Dis 2000; 181 suppl 3: S437-40.
15. Jackson L, Campbell L, Schimidt R, Kuo C, Capuccio A, LeeMJ et al. Specificity of detection of Chlamydia pneumoniaein cardiovascular atheroma. Evaluation of the innocent bystander hypothesis. Am J Pathol 1997; 150(5): 1785-1790.
16. Loopnow H, Libby P, Freudenberg M, Kraus JH, Weckesser J, Mayer H. Citokine induction by lipopolysaccharide (LPS) correspond to lethal toxicity and is inhibited by nontoxic Rhodobacter capsulatus LPS. Infect Immun 1990; 58: 3743-50.
17. Nachtingal M, Legrand A, Greenspan p, Nachtingal SP Nagpal ML. Immortalization of rabbit vascular smooth muscle cells after transfection with a fragment of the Bg1II N region of herpes simplex virus type 2 DNA. Intervirology. 1990; 31: 166-74.
18. O’Connor S. Fulfillment of Koch’s postulates and the causes of atherosclerosis. Am Heart J 1999; 138: s550-1.
19. Fredericks DN, Relman Da. Sequence-based identification of microbial pathogens: a reconsideration of Koch’s postulates. Clin Microbiol Rev 1996; 9: 18-33.
20. Prieto J, Anta L, Alou L, Garcia del Potro M. Macrólidos. En: Antimicrobianos en Medicina. García Sánchez JE, ed Prous Science, Madrid 1999: 349.
21. Gurfinkel E, Bozovich J, Daroca A, Beck E, Mautner B. Randomised trial of roxithromycin in non-Q-wave coronary syndromes: ROXIS Pilot Study. ROXIS Study Group. Lancet 1997; 350: 404-07.
22. Dunne M. WIZARD and the design of trials for secondary prevention of atherosclerosis with antibiotics. Am Heart J 1999; 138: S542-44.
 LA TROMBOSIS UNA COMPLICACIÓN DE LA COVID-19,
LA TROMBOSIS UNA COMPLICACIÓN DE LA COVID-19,
La trombosis puede ser una complicación de la COVID-19, que provoca una «coagulación intravascular diseminada».
Un extenso mensaje que se ha hecho viral a través de WhatsApp alerta de que «a nivel mundial se está atacando mal» la COVID-19, provocada por el coronavirus SARS-CoV-2, ya que, según autopsias realizadas en Italia «no es neumonía, sino coagulación intravascular diseminada» (CID).
El texto, a su vez, tiene su origen en un vídeo en el que el médico hondureño Daniel Dávila Nolasco explica por qué considera que el diagnóstico que se está aplicando contra la Covid-19 es erróneo, una grabación reproducida en algunos medios del país centroamericano y viralizado también en Facebook.
Según esta tesis, compartida múltiples veces durante los últimos días en a través de WhatsApp y redes sociales, «nunca se necesitaron los ventiladores mecánicos» ni las unidades de cuidados intensivos, porque la enfermedad debe tratarse con «antibióticos, antiinflamatorios y anticoagulantes».
El mensaje añade a lo expuesto en el vídeo otros detalles, como un «remedio casero» contra la COVID-19 a base de aspirinas disueltas en zumo de limón hervido y con miel, una supuesta receta milagrosa que también se ha compartido con éxito a través de un video viral en Facebook, acompañado del texto original.
La tesis que presenta la coagulación intravascular como origen del COVID-19 es falsa. porque la trombosis es una complicación que sí se puede provocar como consecuencia de la infección por coronavirus en fases avanzadas, pero es la afectación pulmonar la forma más frecuente de presentación de esta enfermedad.
LA TROMBOSIS PUEDE SER LA CONSECUENCIA FINAL
La coagulación intravascular diseminada (CID) es una complicación que suele manifestarse en los estadios finales de varias situaciones como una infección muy severa, un quemado muy intenso o una sepsis.
«Y, lógicamente, en la Covid-19, con todo lo que provoca, algunos pacientes lo desarrollan; pero no es la causa», explica el presidente de los cardiólogos españoles, antes de insistir: «Al contrario, es la consecuencia final de situaciones que colocan al organismo en un escenario tremendamente complejo».
Así, la coagulación puede ser una consecuencia de la infección por coronavirus y puede que haya «más incidencia de trombosis en sus diversas manifestaciones», pero eso no indica, que se tengan que dejar de utilizar los medicamentos retrovirales. Por el contrario, siguen siendo «fundamentales en los protocolos de tratamiento» del COVID-
Los pacientes que sufren complicaciones graves de la Covid y que son tratados con anticoagulantes tienen la mitad de probabilidad de morir que los que no reciben este tipo de fármacos, según una investigación liderada por el cardiólogo Valentín Fuster en la red de hospitales Mount Sinai de Nueva York.
A raíz de esta investigación, los hospitales neoyorquinos han cambiado la manera de tratar la enfermedad y ahora administran anticoagulantes a todos los pacientes que ingresan por Covid excepto en los casos en que estos fármacos están contraindicados.
“Nunca había visto nada parecido a lo que hace este virus”, declaró ayer en entrevista telefónica Fuster, que dirige el Instituto Cardiovascular de Mount Sinai en EE.UU. y el Centro Nacional de Investigaciones Cardiovasculares (CNIC) en España. “Muchos casos graves tienen un problema importante de exceso de coagulación”.
Este trastorno de coagulación explica los infartos de miocardio, las embolias pulmonares y los ictus que se dan en pacientes de Covid, lo cual llevó a Fuster a pensar que los anticoagulantes podían mejorar el tratamiento de la enfermedad.
“Los fármacos antivirales como el remdesivir son muy importantes para actuar contra el virus, pero no son suficientes para tratar las manifestaciones graves de la infección, que son las que pueden causar la muerte de los pacientes”, sostiene el cardiólogo, que ha organizado la investigación trabajando desde su domicilio en Manhattan.
Para comprobar si los anticoagulantes pueden ser útiles, los médicos de Mount Sinai han analizado en una primera fase del proyecto los datos de 2.773 pacientes ingresados por Covid hasta el 11 de abril. En aquel momento, aún no administraban anticoagulantes a todos los casos de la enfermedad, lo que ha permitido comparar a los que recibieron el tratamiento y a los que no.
Según los resultados presentados ayer en la revista JACC , entre los pacientes conectados a respiradores que no recibieron anticoagulantes, la mortalidad fue del 63%. Entre los que sí recibieron este tipo de fármacos, se redujo al 29%, menos de la mitad. Los porcentajes fueron parecidos para distintos anticoagulantes, tanto inyectados como tomados por vía oral.
Entre los fallecidos, el tiempo medio de permanencia en el hospital antes de morir fue de 9 días sin anticoagulantes y de 21 con el tratamiento. Estos doce días de supervivencia adicional, aunque pueden ser poco relevantes para los pacientes, confirman que el tratamiento tiene una efectividad.
“Uno de los temas que más nos preocupaba era que los anticoagulantes pudieran aumentar el riesgo de hemorragias”, explica Fuster. Pero sólo un 3% de los pacientes que recibieron estos fármacos tuvieron hemorragias, frente al 1,9% de los que no los recibieron. No distraer hiciera
Al tratarse de un estudio retrospectivo, basado en datos de pacientes que recibieron los anticoagulantes en función de la situación en que se encontraba cada uno, los resultados no demuestran que el tratamiento sea la causa de la reducción de mortalidad.
El equipo de Mount Sinai tiene en curso otro estudio retrospectivo con datos de 5.000 pacientes tratados en las últimas cuatro semanas para validar los datos obtenidos hasta ahora. Y tiene previsto iniciar en las próximas semanas dos ensayos clínicos, estos sí prospectivos, para determinar cuál es el mejor anticoagulante y a qué dosis para los pacientes que ingresan en el hospital y para los que requieren cuidados intensivos. Un tercer ensayo clínico comprobará si los anticoagulantes pueden ser útiles para las personas con Covid que no llegan a ingresar en el hospital.
Al mismo tiempo, el equipo de Mount Sinai está investigando con técnicas de biología molecular cómo se origina y cómo progresa el trastorno de coagulación en pacientes con Covid. Todos los datos que estamos acumulando apuntan a que la coagulación desempeña un papel central en las formas graves de esta enfermedad.
Aunque esta línea de investigación está en sus inicios, “hemos decidido publicar los datos que tenemos cuanto antes porque estamos viviendo una situación de emergencia excepcional y estos resultados pueden tener implicaciones importantes para mejorar el tratamiento de los pacientes”.
JOSEP CORBELLA, BARCELONA
07/05/2020 06:00 | Actualizado a 07/05/2020 16:56
Complicaciones de la coagulación
La coagulación de la sangre es uno de los procesos más complejos de nuestro organismo. Cuando la estudiábamos era un tema fascinante y difícil de entender. La finalidad de la coagulación es tratar de evitar la pérdida de sangre cuando hay una herida externa o una lesión interna que provoque una hemorragia. El sistema de reparación es perfecto: primero cerramos el vaso lesionado, lo que hace descender el flujo sanguíneo y favorece la formación de un coágulo. Después, las plaquetas se pegan a las paredes de los vasos y se agregan entre sí. Una vez detenida la hemorragia y reparado el vaso lesionado, el coágulo se disuelve lentamente (fibrinólisis) y volvemos a la normalidad.
Si existe un trastorno de la coagulación pueden formarse coágulos sin lesiones evidentes o los coágulos no llegan a disolverse normalmente. Esta es la situación en enfermedades como la trombosis venosa profunda, el tromboembolismo pulmonar o el accidente vascular cerebral (ictus). Una situación grave es la denominada coagulación intravascular diseminada, en la que se producen coágulos en múltiples vasos sanguíneos de pequeño tamaño que comprometen el flujo de sangre de varios órganos, causando el fallo de algunos de ellos (fallo orgánico múltiple).
Entre un 20% y un 55% de pacientes ingresados en el hospital por padecer la Covid-19 tienen alteraciones analíticas que sugieren la existencia de un trastorno de la coagulación. No sabemos si estas alteraciones son el resultado de la acción directa del virus o un reflejo de las alteraciones inflamatorias que presentan muchos pacientes graves con Covid-19. Tenemos “marcadores” que nos permiten evaluar esta alteración, como el aumento de la concentración del dímero-D (un fragmento de una proteína que se produce cuando un coágulo se disuelve en el interior de nuestro organismo), la prolongación del tiempo de protrombina, el descenso de la cifra de plaquetas o el descenso de los niveles de fibrinógeno. Sabemos ahora que una concentración elevada de dímero-D se asocia a un mayor riesgo de sufrir formas graves de enfermedad y también a mayor mortalidad al comparar estos pacientes con los que tienen niveles normales de dímero-D. Vamos aprendiendo. Aplicación práctica: si no hay contraindicación, a los pacientes con Covid-19 ingresados en los hospitales se les suele administrar profilaxis con anticoagulantes para reducir el riesgo de que desarrollen un tromboembolismo.
ANTONI TRILLA
Hospital Clínic – Universitatde Barcelona – ISGlobal
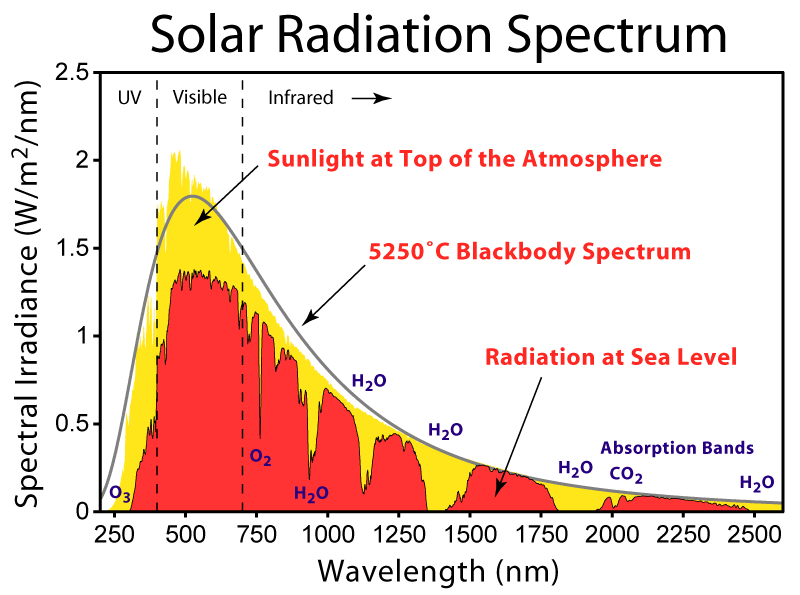 » />>EL CALOR, SOL Y RAYOS ULTRAVIOLETA SON FACTORES NATURALES CONTRA EL CORONAVIRUS
» />>EL CALOR, SOL Y RAYOS ULTRAVIOLETA SON FACTORES NATURALES CONTRA EL CORONAVIRUS
Todas las medidas que se utilizan no son suficiente para controlar y contener el coronavirus COVID- 19, pero la estructura molecular del coronavirus necesita determinadas condiciones ambientales para sobrevivir que se pueden utilizar para luchar contra el
Estas condiciones son características de la estación del año donde se incrementan las temperaturas, el aumento de las horas de luz o los ambientes menos húmedos que en el invierno, parecen ser condiciones que dificultan la propagación de este patógeno que tiene en jaque al sistema sanitario mundial. Al mismo tiempo, en esta época del año, la gente se encuentra durante menos tiempo en espacios cerrados estas estaciones son la primavera y el verano
Con un buen grado de optimismo parece que este coronavirus va a tener una incidencia global y una preciosa los datos parecen indicar que va a ser un virus estacional”, explica a Eltiempo.es José Antonio López Guerrero, profesor titular de microbiología y director del grupo de Neurovirología de la Universidad Autónoma de Madrid (UAM). Y añade: “Este tipo de virus por su estructura molecular, es más sensible a los cambios de temperatura, a la ultravioleta y a la radiación del sol, todas ellas condiciones propias de la primavera y el verano”, .
¿Cuándo desaparecerá el coronavirus?
Además, hay que destacar que con la llegada del calor y la subida de las temperaturas, las personas pasan menos tiempo en espacios cerrados por lo que se reducen las probabilidades de contagio.
“Todo ello tendría que hacer que el virus fuera desapareciendo como han hecho otros virus de características similares antes que este, pero debemos ser prudentes”, advierte este experto ya que a día de hoy todavía hay muchas incógnitas ya que es la primera vez que la sociedad se enfrenta a este virus SARS-COV-2019 que produce la enfermedad COVID-19.
“El virus ahora está en su máxima expansión y hay que ver cómo evoluciona. No obstante, hay que pensar que una gripe en junio o julio es poco probable y algo parecido ocurrirá con este virus”, asegura López.
En esta línea, Rafael M. Ortí Lucas, presidente de la Sociedad Española de Medicina Preventiva y Salud Pública (SEMPSPH) detalla que es probable que la subida de las temperaturas limite la expansión del virus.
Reducción del contagio entre personas
“Como pasa con el virus de la gripe, los coronavirus son virus capsulados, lo que les hace más susceptibles a las condiciones ambientales. En consecuencia , con el calor es previsible que los virus excretados por las mucosas nasales u otras secreciones de una persona afectada se inactiven rápidamente y no lleguen a contagiar a otras personas con tanta facilidad”, comportamiento del virus en la primavera.
Se estima que a temperaturas inferiores a 8ºC exista una mayor supervivencia del virus, e incluso de varios días. “Por contra, temperaturas elevadas, exposición solar y desecación dificultarán su supervivencia en el ambiente, que no deberá pasar de unas pocas horas, y por tanto también el contagio entre personas”, detalla el presidente de la SEMPSPH.
No obstante, hay que tener en cuenta que los expertos no han realizado ninguna predicción sobre este novedoso virus por lo que las respuestas que facilitan se basan en el comportamiento de virus similares estudiados con anterioridad.
 QUE SON ONDAS ELECTROMAGNÉTICAS
QUE SON ONDAS ELECTROMAGNÉTICAS
Son aquellas ondas que no necesitan un medio material para propagarse. Incluyen, entre otras, la luz visible y las ondas de radio, televisión y telefonía.
Todas se propagan en el vacío a una velocidad constante, muy alta (300 0000 km/s) pero no infinita. Gracias a ello podemos observar la luz emitida por una estrella lejana hace tanto tiempo que quizás esa estrella haya desaparecido ya. O enterarnos de un suceso que ocurre a miles de kilómetros prácticamente en el instante de producirse.
Las ondas electromagnéticas se propagan mediante una oscilación de campos eléctricos y magnéticos. Los campos electromagnéticos al «excitar» los electrones de nuestra retina, nos comunican con el exterior y permiten que nuestro cerebro «construya» el escenario del mundo en que estamos.
Las O.E.M. son también soporte de las telecomunicaciones y el funcionamiento complejo del mundo actual.
Las cargas eléctricas al ser aceleradas originan ondas electromagnéticas
El campo E originado por la carga acelerada depende de la distancia a la carga, la aceleración de la carga y del seno del ángulo que forma la dirección de aceleración de la carga y al dirección al punto en que medimos el campo( sen ).
Un campo electrico variable engendra un campo magnético variable y este a su vez uno electrico, de esta forma las o. e.m. se propagan en el vacio sin soporte material
CARACTERÍSTICAS de LA RADIACIÓN E.M.
Los campos producidos por las cargas en movimiento puden abandonar las fuentes y viajar a través del espacio ( en el vacio) creándose y recreándose mutuamente. Lo explica la tercera y cuarta ley de Maxwell.
Las radiaciones electromagnéticas se propagan en el vacio a la velocidad de la luz «c». Y justo el valor de la velocidad de la luz se deduce de las ecuaciones de Maxwell, se halla a partir de dos constantes del medio en que se propaga para las ondas electricas y magnética .
Los campos electricos y magnéticos son perpendiculares entre si ( y perpendiculares a la dirección de propagación) y estan en fase: alcanzan sus valores máximos y mínmos al mismo tiempo y su relación en todo momento está dada por E=c· B
El campo eléctrico procedente de un dipolo está contenido en el plano formado por el eje del dipolo y la dirección de propagación. El enunciado anterior también se cumple si sustituimos el eje del dipolo por la dirección de movimiento de una carga acelerada
Las ondas electromagnéticas son todas semejantes ( independientemente de como se formen) y sólo se diferencian e n su longitud de onda y frecuencia. La luz es una onda electromagnética
Las ondas electromagnéticas transmiten energía incluso en el vacio. Lo que vibra a su paso son los campos eléctricos y magnéticos que crean a propagarse. La vibracion puede ser captada y esa energía absorberse.
Las intensidad instantánea que posee una onda electromagnética, es decir, la energía que por unidad de tiempo atraviesa la unidad de superficie, colocada perpendicularmente a la direción de propagación es: I=c· E2. La intensidad media que se propaga es justo la mitad de la expresión anterior.
La intensidad de la onda electromagnética al espandirse en el espacio disminuuye con el cuadrado de la distancia y como «I «es proporcional a E2 y por tanto a sen2 . Por lo tanto existen direcciones preferenciales de propagación
ESPECTRO ELECTROMAGNÉTICO
Las ondas electromagnéticas se agrupan según su frecuencia, aunque no existe un límte muy presiso para cada grupo.
Una misma fuente de ondas electromagfnéticas puede generar al mismo tiempo ondas de varios tipos.
Ondas de radio: son las utilizadas en telecomunicaciones e incluyen las ondas de radio y televisión. Su frecuencia oscila desde unos pocos hercios hasta mil millones de hercios. Se originan en la oscilación de la carga eléctrica en las antenas emisoras (dipolo radiantes).
Microondas: Se utilizan en las comunicaciones del radar o la banda UHF ( Ultra High Frecuency) y en los hornos de las cocinas. Su frecuencia va desde los milmillones de hercios hasta casi el billon.Se producen en oscilaciones dentro de un aparato llamado magnetrón. El magnetrón es una cavidad resonante formada por dos imanes de disco en los extremos, donde los electrones emitidos por un cátodo son acelerados originado los campos electromagnéticos oscilantes de la frecuencia de microondas.
Infrarrojos: Son emitidos por los cuerpos calientes. Los transitos energéticos implicados en rotaciones y vibraciones de las moléculas caen dentro de este rango de frecuencias. Los visores nocturnos detectan la radiación emitida por los cuerpos a una temperatura de 37 º .Sus frecuencias van desde 10 11Hz a 4·1014Hz. Nuestra piel también detecta el calor y por lo tanto las radiaciones infrarrojas.
Luz visible: Incluye una franja estrecha de frecuencias, los humanos tenemos unos sensores para detectarla ( los ojos, retina, conos y bastones). Se originan en la aceleración de los electrones en los tránsitos energéticos entre órbitas permitidas. Entre 4·1014Hz y 8·1014Hz
Ultravioleta: Comprende de 8·1014Hz a 1·1017Hz. Son producidas por saltos de electrones en átomos y molécualas excitados. Tiene el rango de energía que interviene en las reacciones químicas. El sol es una fuente poderosa de UVA ( rayos ultravioleta) los cuales al interaccionar con la atmósfera exterior la ionizan creando la ionosfera. Los ultravioleta puden destruir la vida y se emplean para esterilizar. Nuestra piel detecta la radiación ultravioleta y nuestro organismo se pone a fabricar melanina para protegernos de la radiación. La capa de ozono nos proteje de los UVA.
Rayos X: Son producidos por electrones que saltan de órbitas internas en átomos pesados. Sus frecuencias van de 1’1·1017Hz a 1,1·1019Hz. Son peligrosos para la vida: una exposición prolongada produce cancer.
Rayos gamma: comprenden frecuencias mayores de 1·1019Hz. Se origina en los procesos de estabilización en el núcleo del átomo después de emisiones radiactivas. Sus radiación es muy peligrosa para los seres vivos.
CAMPOS ELECTROMAGNÉTICOS Y EFECTOS BIOLÓGICOS
En los tiempos de virus que corren, es infrecuente hablar de los efectos nocivos que tienen las radiaciones Solares sobre los virus
Cada día es muy frecuente en los medios de comunicación hablar de vacunas para el virus concretamente para el Covid 19 y otra serie de tratamientos médicos que de una manera ansiosa buscamos para controlar esta epidemia que nos está asolando
desde muy antiguo se conocen por sus efectos nocivos de la radiaciones sobre virus entre otros gérmenes.
Siempre digo que pocas veces he visto una gripe en verano. Algo pasa en verano que mutila a los virus y no les deja actuar
La energía electromagnética es emitida en forma de ondas por las fuentes naturales y por numerosas fuentes artificiales. Esas ondas consisten en campos eléctricos y magnéticos oscilantes que se influyen recíprocamente y de diferentes formas con sistemas biológicos tales como células, plantas, animales o seres humanos. Para comprender mejor esa influencia recíproca, es indispensable conocer las propiedades físicas de las ondas que constituyen el espectro magnético.
Las ondas electromagnéticas pueden caracterizarse por su longitud, frecuencia y energía. Los tres parámetros se relacionan entre sí. Cada uno de ellos condiciona el efecto del campo sobre un sistema biológico.
La frecuencia de una onda electromagnética es en definitiva el número de veces que cambia el sentido del campo en la unidad de tiempo en un punto dado. Se mide en ciclos por segundo, o Hercios.
Cuanto más corta es la longitud de onda, más alta es la frecuencia. Por ejemplo, el tramo intermedio de una banda de radiodifusión de amplitud modulada tiene una frecuencia de un millón de herzios (1 Mhz) y una longitud de onda de aproximadamente 300 metros. Los hornos de microondas utilizan una frecuencia de 2.450 millones de Herzios (2,45 Ghz) y tienen una longitud de onda de 12 centímetros.
Una onda electromagnética está formada por paquetes muy pequeños de energía llamados fotones. La energía de cada paquete o fotón es directamente proporcional a la frecuencia de la onda: Cuanta más alta es la frecuencia, mayor es la cantidad de energía contenida en cada fotón.
El efecto de las ondas electromagnéticas en los sistemas biológicos está determinado en parte por la intensidad del campo y en parte por la cantidad de energía contenida en cada fotón.
Las ondas electromagnéticas de baja frecuencia se denominan «campos electromagnéticos», y las de muy alta frecuencia, «radiaciones electromagnéticas». Según sea su frecuencia y energía, las ondas electromagnéticas pueden clasificarse en «radiaciones ionizantes» o «radiaciones no ionizantes».
Las radiaciones ionizantes son ondas electromagnéticas de frecuencia extremadamente elevada (rayos X y gamma), que contienen energía suficiente para producir la ionización (conversión de átomos o partes de moléculas en iones con carga eléctrica positiva o negativa) mediante la ruptura de los enlaces atómicos que mantienen unidas las moléculas en la célula.
Las radiaciones no ionizantes constituyen, en general, la parte del espectro electromagnético cuya energía es demasiado débil para romper enlaces atómicos. Entre ellas cabe citar la radiación ultravioleta, la luz visible, la radiación infrarroja, los campos de radiofrecuencias y microondas, los campos de frecuencias extremadamente bajas y los campos eléctricos y magnéticos estáticos.
Las radiaciones no ionizantes, aún cuando sean de alta intensidad, no pueden causar ionización en un sistema biológico. Sin embargo, se ha comprobado que esas radiaciones producen otros efectos biológicos, como por ejemplo calentamiento, alteración de las reacciones químicas o inducción de corrientes eléctricas en los tejidos y las células.
Las ondas electromagnéticas pueden producir efectos biológicos que a veces, pero no siempre, resultan perjudiciales para la salud. Es importante comprender la diferencia entre ambos:
Un efecto biológico se produce cuando la exposición a las ondas electromagnéticas provoca algún cambio fisiológico perceptible o detectable en un sistema biológico.
Un efecto perjudicial para la salud tiene lugar cuando el efecto biológico sobrepasa la capacidad normal de compensación del organismo y origina algún proceso patológico.
Algunos efectos biológicos pueden ser inocuos, como por ejemplo la reacción orgánica de incremento del riego sanguíneo cutáneo en respuesta a un ligero calentamiento producido por el sol. Algunos efectos pueden ser provechosos, como por ejemplo la sensación cálida de la luz solar directa en un día frío, o incluso beneficiosos para la salud, como es el caso de la función solar en la producción de vitamina D por el organismo. Sin embargo, otros efectos biológicos, como son las quemaduras solares o el cáncer de piel, resultan perjudiciales para la salud.
Es sabido que los campos de radiofrecuencias producen calentamiento e inducen corrientes eléctricas. Asimismo, se han notificado otros efectos biológicos menos probados.
Los campos de radiofrecuencias de frecuencia superior a 1 Mhz causan sobre todo calentamiento, al desplazar iones y moléculas de agua a través del medio al que éstos pertenecen. Incluso a niveles muy bajos, la energía de las radiofrecuencias produce pequeñas cantidades de calor, que son absorbidas por los procesos termorreguladores normales del organismo sin que el individuo lo perciba.
Los campos de radiofrecuencias de frecuencia inferior a 1 Mhz aproximadamente inducen principalmente cargas y corrientes eléctricas que pueden estimular células de tejidos tales como los nervios y los músculos. Las corrientes eléctricas están ya presentes en el organismo como parte normal de las reacciones químicas propias de la vida. Si los campos de radiofrecuencias inducen corrientes que excedan significativamente ese nivel de base en el organismo, es posible que se produzcan efectos perjudiciales para la salud.
Campos eléctricos y magnéticos de frecuencias extremadamente bajas: La acción primordial de estos campos en los sistemas biológicos es la inducción de cargas y corrientes eléctricas. Es poco probable que esa acción baste para explicar efectos sanitarios tales como el cáncer infantil, que se ha notificado como causado por la exposición a niveles «ambientales» de campos de frecuencias extremadamente bajas.
Campos eléctricos y magnéticos estáticos. Aunque la acción principal ejercida por esos campos en los sistemas biológicos es la inducción de cargas y corrientes eléctricas, se ha comprobado la existencia de otros efectos que, en principio, podrían resultar perjudiciales para la salud, pero sólo en campos de intensidades muy elevadas.
Los campos eléctricos estáticos no penetran en el organismo tanto como los campos magnéticos, pero pueden percibirse por el movimiento del vello cutáneo. Aparte de las descargas eléctricas de campos electrostáticos potentes, no parecen tener efectos apreciables para la salud.
Los campos magnéticos estáticos tienen prácticamente la misma intensidad dentro del cuerpo que fuera de él. Cuando esos campos son muy intensos, pueden alterar el riego sanguíneo o modificar los impulsos nerviosos normales. Pero inducciones magnéticas tan elevadas no se producen en la vida diaria. Ahora bien, no se dispone de suficiente información sobre los efectos de la exposición duradera a campos magnéticos estáticos a los niveles existentes en el entorno laboral.
Con objeto de asegurar que la exposición humana a los campos electromagnéticos no tenga efectos perjudiciales para la salud, que los aparatos generadores de esos campos sean inocuos y que su utilización no cause interferencias eléctricas con otros aparatos, se han adoptado diversas directrices y normas internacionales. Esas normas se elaboran después de que grupos de científicos, que buscan pruebas de la repetición sistemática de efectos perjudiciales para la salud, hayan analizado todas las publicaciones científicas. Posteriormente, esos grupos recomiendan directrices que permitirán a los órganos nacionales e internacionales correspondientes preparar normas prácticas. La Comisión Internacional de Protección contra las Radiaciones No Ionizantes (ICNIRP), organización no gubernamental reconocida oficialmente por la OMS en el sector de la protección contra las radiaciones no ionizantes, ha establecido directrices internacionales sobre los límites de la exposición humana para todos los campos electromagnéticos, con inclusión de la radiación ultravioleta, la luz visible y la radiación infrarroja.
La interacción de las ondas electromagnéticas y los sistemas biológicos, tales como células, plantas, animales o seres humanos, difiere en función de la frecuencia de esas ondas. La medida en que tales ondas afectan a los sistemas biológicos depende en parte de su intensidad y en parte de la cantidad de energía (de la frecuencia) Los efectos biológicos pueden, en ocasiones, pero no siempre, resultar perjudiciales para la salud
La carga viral del COVID 19 es un marcador útil para evaluar la gravedad y el pronóstico
Los pacientes graves con COVID-19 tienden a tener una alta carga viral y un largo período de eliminación del virus
Federico Pérez
23 de marzo 2020. 3:10 pm
La dinámica viral en casos leves y graves de COVID 19 ha sido el objeto de estudio de un grupo de investigadores del Hospital of Nanchang University que publican los resultados en The Lancet Infectious Diseases. El estudio concluye que la carga viral de SARS-CoV-2 podría ser un marcador útil para evaluar la gravedad y el pronóstico de la enfermedad
Anteriormente estos autores ya habían realizado estudios del mismo tipo en pacientes con Síndrome Respiratorio Agudo (SARS) encontrando que la carga viral del coronavirus del síndrome respiratorio agudo severo (SARS-CoV-2) alcanza su punto máximo dentro de la primera semana del inicio de la enfermedad.
Los resultados de febrero de 2020 indicaron que el espectro clínico de esta enfermedad puede ser muy heterogéneo.
En este nievo estudio, Yang Liu, y su equipo de colaboradores investigan la dinámica viral analizando los patrones de ARN viral observados en pacientes con COVID-19 leve y grave.
Los autores incluyeron en el estudio 76 pacientes ingresados en el Primer Hospital Afiliado de la Universidad de Nanchang (Nanchang, China) del 21 de enero al 4 de febrero de 2020. Se confirmó que todos los pacientes tenían COVID-19 en el momento del ingreso por determinación con RT-PCR, y se analizaron las cargas virales obtenidas de muestras recogidas con torunda nasofaríngea.
Liu indica que se clasificó a los pacientes como “casos graves” aquellos que tenían cualquiera de las siguientes características en el momento de la admisión: dificultad respiratoria (≥30 respiraciones por minuto); saturación de oxígeno en reposo ≤93%; relación entre la presión parcial de oxígeno arterial y la concentración parcial de aire inspirado en oxígeno ≤300 mm Hg; o complicaciones graves de la enfermedad (p. ej., insuficiencia respiratoria, necesidad de ventilación mecánica, shock séptico o insuficiencia orgánica no respiratoria).
46 (61%) individuos fueron clasificados como casos leves y 30 (39%) fueron clasificados como casos severos. Los datos demográficos básicos y los síntomas clínicos iniciales de estos pacientes no difirieron significativamente entre los grupos, excepto que los pacientes en el grupo severo eran significativamente mayores que los del grupo leve, como se esperaba, señalan los investigadores
Ningún paciente murió por la infección. El 77 por ciento de los casos graves recibieron tratamiento en la unidad de cuidados intensivos (UCI), mientras que ninguno de los casos leves requirió tratamiento en la UCI.
Todas las muestras fueron recolectadas de acuerdo con las pautas de la OMS.
La carga viral media de los casos graves fue alrededor de 60 veces mayor que la de los casos leves, lo que sugiere que las cargas virales más altas podrían estar asociadas con resultados clínicos graves.
Liu y su equipo también estudiaron muestras en serie de 21 casos leves y diez casos graves, encontrando que los casos leves tenían un aclaramiento viral temprano, con el 90% de estos pacientes que dieron repetidamente resultados negativos en RT-PCR al cabo de 10 después del inicio. Por el contrario, todos los casos graves todavía dieron positivo en el día 10 después del inicio. “En general, nuestros datos indican que es similar al SARS en 2002–03” según los autores.
Los pacientes con COVID-19 grave tienden a tener una alta carga viral y un largo período de eliminación del virus. Este hallazgo sugiere que la carga viral de SARS-CoV-2 podría ser un marcador útil para evaluar la gravedad y el pronóstico de la enfermedad.
 >CORONAVIRUS, INMUNOPATOGENIA VIRAL
>CORONAVIRUS, INMUNOPATOGENIA VIRAL
.
los mecanismos que conducen a la inmunología y la patogenia del nuevo coronavirus SARS-CoV-2 son complejos
Un virus es capaz de penetrar a través de las barreras físicas que forma nuestro recubrimiento epitelial o mucoso, por ejemplo, en el tracto respiratorio. Y que es capaz de resistir las barreras químicas y biológicas creadas por nuestros microorganismos comensales, las células epiteliales y por las células inmunitarias que producen desagradables
ambientes ácidos, grasos y salinos o que secretan enzimas tóxicos o péptidos catiónicos que podrían destruir sus biomoléculas y con ello su integridad.
Si el virus ha sido capaz de saltarse esta barrera de elementos defensivos constitutivos y preformados ya lo tenemos en nuestro interior. Nos ha infectado. Ahora necesitamos que nuestras células sean capaces de percibirlo, que puedan iniciar la respuesta inmunitaria innata inducida y la inflamatoria con el objetivo de contenerlo y erradicarlo en menos de 96 horas. Pero en este contexto hay que tener en cuenta un detalle muy importante, el virus además de penetrar ha de dañar discretamente nuestros tejidos. Ello no debería ser ningún obstáculo para él, pues seguramente tendrá tropismo por alguna célula del epitelio, penetrará así en su interior, le secuestrará la maquinaria biosintética en su propio beneficio y generará nuevos viriones con el consiguiente estallido y necrosis de la célula infectada.
La detección de la infección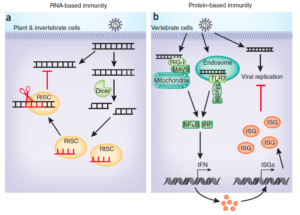
Con estas muertes celulares no programadas se van a liberan de forma masiva moléculas endógenas celulares que además van a dañar o modifica el andamiaje de los tejidos. Estas moléculas o cambios los denominamos patrones moleculares asociados al daño (también conocidos como DAMP o alarminas) y son inmunogénicos pues las células de la inmunidad innata poseen receptores que van a ser capaces de percibirlos y reconocerlos.
Pero no debemos olvidar que además del daño tenemos al extraño, al virus que ha conseguido infectarnos. Pues bien, los microorganismos patogénicos también tienen otros patrones moleculares asociados (en términos inmunológicos PAMP o MAMP) que serán reconocidos por los sensores celulares. Como los microorganismos pueden localizarse o esconderse en cualquier ubicación del individuo, sus células y especialmente las del sistema inmunitario, disponen de un gran abanico de sensores de estos patrones y los denominamos receptores que reconocen patrones (PRR). Estos numerosos PRRs pertenecen a distintas familias estructurales y así mismo cumplen funciones muy diferentes. Hay sensores que se localizan tanto en las membranas de la célula (por ejemplo, toll-like receptors o TLR, scavenger receptors o SR y los receptores de manosa de los macrófagos o MMR) como en el citoplasma de la misma (los NOD-like receptors o NLR y los RIG-like receptors o RLR), como en los humores o fluidos biológicos (la lectina que une manosa o MBL y la proteína C-reactiva o CRP). Este último tipo de PRR humorales los producen también otras células como los hepatocitos, que los liberan a la sangre para que se repartan por todo el organismo.
Estos PRRs además de la función como sensor cumplen otras dos funciones muy distintas y a la vez muy importantes para la defensa del individuo. Por un lado, la fagocitosis de los microorganismos que sean reconocidos por los neutrófilos y los macrófagos y, por otro lado, la activación del complemento, el principal ejecutor de la inmunidad innata humoral. Esta función tipo sensor va a disparar la respuesta inmunitaria innata local, la respuesta inflamatoria y la de reparación tisular. Se va a aumentar la permeabilidad de los vasos para que nuevos componentes del sistema inmunitario puedan acudir a este territorio que está siendo invadido por los virus y que la suma y la unión haga la fuerza antimicrobiana. También es muy destacable tener en cuenta que esta respuesta inflamatoria y la actividad de las células dendríticas y macrófagos presentando antígenos y estimulando a los linfocitos T, también va a llevar a la activación de la respuesta inmunitaria adaptativa. Esta última es la que en última instancia combatirá de la forma más específica, especializada y efectiva la infección del virus, este parásito intracelular y además podremos beneficiarnos de su efecto memoria ante futuras exposiciones del mismo microbio.
La respuesta antiviral intrínseca
Volvamos otra vez a fijarnos en el agente invasor, el virus, el cual debido a que es una partícula no viva y de absoluta simplicidad estructural podría ser muy resistente y esquivo a todas estas formas de reconocimiento y medidas que la respuesta inmunitaria esta generando contra el mismo. Pero nuestras células y nuestro sistema inmunitario han evolucionado a lo largo de millones de años de forma muy satisfactoria, conocedores de que existen los virus y que para poder detectarlos y contrarrestarlos son necesarias otras estrategias muy diferentes que las que generamos contra los otros tipos de microorganismos.
Yan N, Nat Immunol 2012
Y nos estamos refiriendo a la inmunidad antiviral intrínseca que restringe directamente la replicación viral y el ensamblaje, lo que hace que una célula no sea permisiva a la colonización viral. Esta inmunidad intrínseca es conferida por factores preexistentes y en gran medida por la presencia de factores inducidos por la propia infección del virus. Una de las formas más simples de impedir la actividad y replicación viral se observa en invertebrados y la denominamos inmunidad basada en el RNA al utiliza distintos tipos de RNA de interferencia que bloquearán la expresión de los genes virales e incluso inducirán su degradación. Este sistema es comparable a la regulación epigenética que realizan los microRNAs usando las enzimas Dicer y RISC o de una forma análoga al conocido sistema antiviral bacteriano CRISPR-Cas. Aumentando la complejidad evolutiva, los vertebrados hemos desarrollado otro mecanismo más complejo y eficiente, que es una inmunidad basada en proteínas y es la respuesta a los interferones (su nombre deriva de que interfiere en la capacidad infectiva de un virus). Como vamos a ver, la respuesta del interferón que desencadena alguno de los citados PRRs al detectar un virus no solo le afecta a la célula que percibe este invasor (autocrina), sino que se extenderá de forma paracrina a las células de su vecindario para que todas puedan degradar la información genética del virus e impidan la expresión de sus genes.
La respuesta del interferón
Llegado este momento es importante que veamos de forma algo más detallado en qué consiste la respuesta antiviral mediada por interferón. Los principales PRR que detectan la presencia de virus se basan en localizar ácidos nucleicos no “naturales” de la célula como dsRNA y DNA con CpG no metilados, y se localizan en las membranas de las vesículas endocíticas (dentro de la célula) y son los TLR3, 7, 8 y 9, además de los RLR citosólicos RIG-I y MDA5. Mediante un mecanismo complejo de señalización y transducción estos PRR consiguen que se fosforilen y dimericen componentes de una familia de factores de transcripción que se denominan IRF (interferon regulatory factor) y que puedan translocarse al núcleo. Una vez en el núcleo, estos IRF interaccionaran con genes que contienen en sus promotores secuencias nucleotídicas denominadas ISRE (IFN-stimulated response elements) y que conducen a la expresión de los interferones de tipo I (INF-a e IFN-b) y otros genes antivirales importantes (que ahora luego volveremos con ellos).
Gilliet M, Nat Rev immune 2008
Hay que hacer notar que este interferón que va a producir la célula no interfiere directamente con el virus, sino que va a ser secretado y las células que posean receptores para los interferones de tipo I (IFNAR) serán las que se beneficien de su producción y liberación. Afortunadamente la expresión del receptor de IFN se transcribe en ese contexto de ISRE por lo que la célula productora será una de las beneficiadas junto con otras células del microambiente. La señalización a través de los IFNAR lleva a un mecanismo similar donde intervienen otros factores de transcripción que actúan sobre las mismas secuencias ISRE y alimentan como un bucle con feedback positivo todo el proceso de señalización y protección local contra los virus.
Michalska A, Front Immunol 2018
Nos queda ver ahora cuales esas proteínas que se van a producir gracias a los interferones y son la que directamente van en contra de los virus. En primer lugar, se aumenta la expresión del complejo principal de histocompatibilidad de tipo I y la presentación de proteínas celulares (y también virales, pues el virus está dentro de la célula y usa su maquinaria metabólica) a los linfocitos T. Este proceso fundamental para la vigilancia inmunológica permite que los linfocitos detecten la producción de proteínas extrañas al organismo (como las virales) y si se da el caso, que se genere la respuesta inmunitaria adaptativa. Todo ello acompañado en un contexto que maximiza la activación de los linfocitos citotóxicos (CTL) que en última instancia serán los encargados de destruir a células infectadas por virus. Además de la activación de los CTL, la célula produce una serie de enzimas que degradan RNAs virales como la 2′,5′ oligoadenilato sintetasa, PKR que impiden la acción de factores de transcripción necesarios para la expresión de proteínas virales y proteasas que degradan las proteínas de la cápside viral que se hayan sintetizado, impidiendo el ensamblaje de nuevos viriones. De esta forma tan eficiente la inmunidad innata logra detectar los virus y no solo luchar contra ellos, sino que iniciar la activación de la respuesta inmunitaria adaptativa.
Abbas A, Cellular and Molecular Immunology, Elsevier
Pero los virus no iban a ser menos que las células de los vertebrados y su proceso evolutivo a algunos también les ha permitido adquirir mecanismos de resistencia o de evasión de la respuesta antiviral y por ende de la respuesta inmunitaria contribuyendo a la inmunopatogenia de algunas enfermedades virales. Estas estrategias son muy variadas y están asociadas a la complejidad genómica del virus. Cuantos más genes sean capaces de empaquetar en sus cápsides, seguramente más genes tendrán para bloquear la señalización de PRR y de IFNAR. Para poder entenderlo mejor, lo más conveniente será ver un ejemplo y en estos momentos tal vez el ejemplo más interesante es el coronavirus. Vayamos pues a ver los mecanismos inmunopatogénicos de los coronavirus
Un ejemplo concreto, el Coronavirus y la epidemia Covid-19
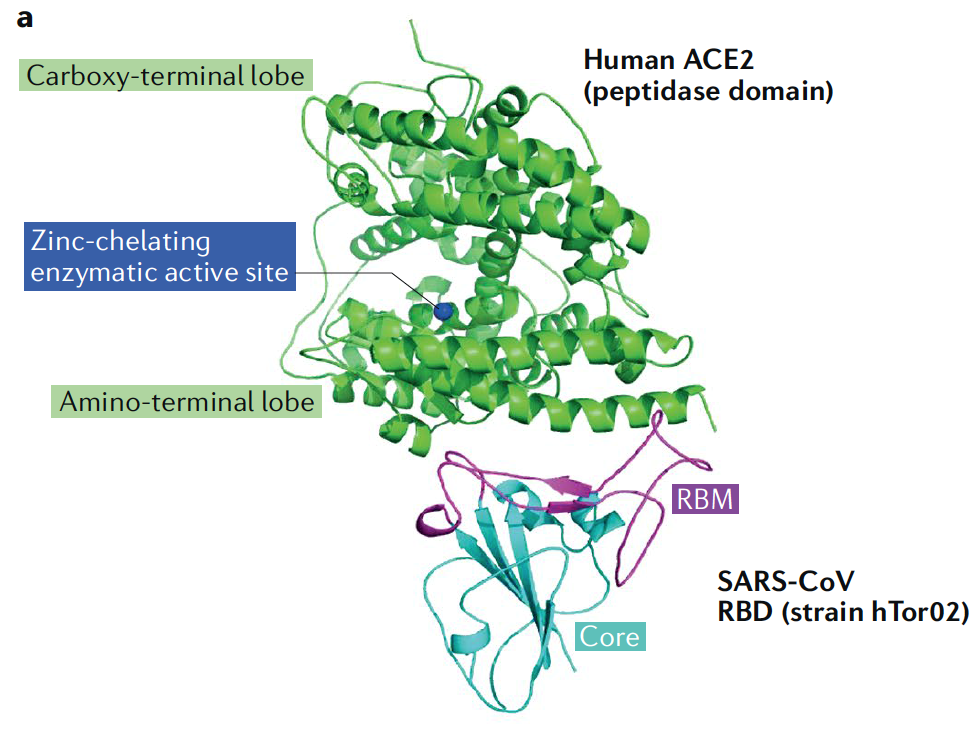
En diciembre del año 2019 la humanidad ha sufrido por tercera vez en las últimas dos décadas una nueva zoonosis causada por un coronavirus (SARS-CoV-2), provocando una epidemia que afecta a las vías respiratorias y que en algunos caos es una enfermedad muy grave, la denominada Covid-19. El avance la biología molecular y el riesgo a que se convierta en una pandemia ha intensificado los esfuerzos de los centros de investigación, las autoridades sanitarias de muchos países y de la OMS. Gracias a ello, en tan poco tiempo tenemos muchos datos epidemiológicos, conocemos la información genética del virus y se han desarrollado test diagnósticos muy sensibles y cuantitativos. Se ha podido establecer el tropismo viral sobre células epiteliales alveolares gracias a la interacción con la exopeptidasa, enzima convertidora de angiotensina (hACE2). Esta enzima transforma, preferentemente en el pulmón, al precursor de la angiotensina en una molécula activa y que se encarga de regular la presión arterial. Pero aún quedan muchas lagunas por resolver y que nos pueden ayudar a contener la epidemia, como cuál ha sido el vector, el desarrollo de algún antiviral o de una vacuna. A pesar de todo, el esfuerzo tecnológico y humano ha sido tremendo y sin parangón.
Cui J, Nat Rev Microbiol 2019
El virus y sus genes estructurales
Los Coronavirus son virus de RNA monocatenario con capacidad codificante (+ssRNA) y en este caso concreto, el tamaño del genoma de SARS-CoV-2 es de 30 kb. Teniendo en cuenta que 10 kb es el tamaño medio de los virus de RNA, este detalle sitúa a los coronavirus como los virus de RNA con genoma mas grande.
Banerjee A, Curr Clin Micro Rpt (2019)
Y tener un genoma mayor conlleva mejoras en su supervivencia y mejoras en su adaptación al hospedador lo que implica en muchos casos capacidad de escape inmunológico o cierta inmunopatogenia. Esta complejidad, en primer lugar, se asocia con una característica muy relevante a la hora de la multiplicación y la evolución viral y es que el complejo de replicación transcripción tiene actividad 3’‐5’ exorribonucleasa, lo que le confiere mayor fidelidad de copia. Luego los coronavirus mutan, seguro que mucho, pero menos de lo que cabría esperar tiendo en cuenta que es un virus de RNA. Las proteínas que están codificadas en su genoma se dividen en dos grupos, proteínas no estructurales y proteínas estructurales.
Chen Y, J Med Virol 2020
Empecemos por estas últimas, y dentro de las estructurales, en sentido 5’-3’ tenemos:
La proteína S (Spike, pincho), que formando homotrímeros crea esas estructuras que asoman sobre la nuceocápside y le permiten anclarse a su receptor (hACE2).
La proteína E (Envuelta) encargada del ensamblaje viral y la patogenia del virus.
La proteína M (Membrana) tiene tres dominios transmembrana y al unirse a la nucleocápside le confiere la estructura esférica.
La proteína N (Nucleocápside) tiene dos dominios, uno que interacciona con el RNA viral conduciendo a su correcto encapsulamiento en los viriones y otro dominio multifunción que actúa por un lado como antagonista del IFN y por otro lado como represor de los RNA de interferencia, lo cual es muy importante para evitar la acción inmunitaria antiviral y es beneficios para su replicación.
Además, los b-Coronavirus también disponen de la hemaglutinina-acetilesterasa (HE), una glicoproteína que se une a restos de azúcar en las membranas celulares e induce la fusión y penetración viral en la célula. Curiosamente, parece ser que el gen para HE fue introducido en un genoma ancestral de coronavirus por recombinación con el virus de la influenza C.
Holmes KV, N Engl J Med 2003
Los genes no estructurales y la inmunopatogenia viral
El análisis genético y molecular del SARS-CoV-2 nos permite afirmar que no es ni un mutante de cualquier coronavirus anterior ni un recombinante de coronavirus conocidos. Es un coronavirus desconocido, probablemente de un hospedador no humano que de alguna manera adquirió la capacidad de infectar a humanos y provocar la presente zoonosis.
Zhu N, N Engl J Med 2020
Sobre los genes no estructurales empezaremos abordando el papel que juegan las otras proteínas virales, así:
La nsp13 es una helicasa, una enzima vital para el desempaquetado de los genes y por ende la replicación y transcripción.
Las nsp7 a 10 son regiones críticas como reguladoras de la nsp12, que es la RNA polimerasa multidominio y que junto con la actividad de reparación del RNA (MMR) de la nsp14 le confiere estas altas tasas de fidelidad de copia.
Por otro lado, como la síntesis del mRNA del coronavirus se realiza en el citosol en lugar del núcleo, los virus de +RNA de eucariotas no pueden confiar en que las proteínas nucleares de la célula se encarguen de añadir la 7-metilguanosina (m7G) al primer nucleótido del RNA para protegerlo de las exorribonucleasas celulares (proceso que se denomina capping) y conducir a su traducción en proteínas. En este sentido, las nsp10-13-14-16 virales participan en el capping del RNA viral.
Veamos entonces las claves de la inmunopatogenia de los coronavirus que pivotan sobre el bloque de la actividad antiviral, que se pone de manifiesto gracias a la acción del resto de proteínas no estructurales:
La nsp1 de los SARS-CoV es una proteína de 20 kDa que se localiza en el citoplasma de las células infectadas y que genera una variedad de funciones únicas en los b-Coronavirus. En primer lugar, es capaz de bloquear y degradar el mRNA del IFN-b y otros mensajeros endógenos. En segundo lugar, al bloquear la fosforilación de STAT1 e I-kB y la dimerización de IRF3, inhibe las vías de transducción de señales que involucran IRF, STAT y NF-k Ello afecta también a la progresión del ciclo celular sin inducir apoptosis, favoreciendo la supervivencia viral.
La nsp3, codificada en el gran ORF1 y que genera una proteína de 200 kDa con varios dominios funcionales diferentes. Estos incluyen un dominio de unión de poli (ADP-ribosa), de proteasa (papain-like protease, PLP), otro deubiquitinasa y un último de ISGylase. La función PLP de nsp3 es la que antagoniza las respuestas IFN de tipo I, impidiendo la fosforilación, dimerización e importe al núcleo de IRF3. De forma análoga actúa sobre la vía de represión de NF-k Además, la función ISGylase contrarresta la acción del gen 15 estimulado por interferón (ISG15), que es la ISGilación. Veamos en qué consiste este proceso. La modificación postraduccional de las proteínas es una estrategia importante para la regulación del proteoma celular. La ubiquitina y otros modificadores similares a la ubiquitina como ISG15, a través de su conjugación covalente a proteínas median la regulación de los niveles de esas proteínas, las vías de señalización, el tráfico vesicular y muchos otros procesos celulares. ISG15 se codifica y se expresa como una proteína precursora de 17 kDa que se procesa proteolíticamente en su extremo C-ter para exponer una secuencia de aminoácidos, idéntica a la secuencia C-ter de la ubiquitina, y sus adiciones covalentes a las proteínas son bastante similares. Por este mecanismo se regula a la baja la vida media de las proteínas virales, disminuyendo su capacidad de ensamblaje y la viabilidad de los mismos. De forma contraria este proceso afecta aumentando la vida media de muchos genes regulados al alza como respuesta de IRF. Por este motivo esta actividad ISGylase parece una estrategia acertada de propagación viral.
Morales DJ, J Mol Biol 2013
Las otras nsp4 a nsp6 bloquean directamente la actividad antiviral que la célula debería poner en marcha inhibiendo la señalización del IFN. Algunas formas descritas por SARS-CoVs de antagonizar a los IFN de tipo I es mediante la inactivación de la traducción del hospedador; la degradación de sus mARN; interactuar negativamente con STING; antagonizar la señalización de IFN inducida por MAVS/RIG-I. Así mismo, también lo consiguen bloqueando la expresión de citocinas proinflamatorias y la generación de las vesículas que son necesarias para la formación de autofagosomas y el incremento de la presentación de péptidos virales.
Por último, la nsp16 es una 2’O metiltransferasa (2’‐O‐MTasa) que bloquea muy selectivamente la señalización a través de MDA5, un tipo de RIG-like receptor.
Tortura AL, Curr Opinion Virol 2012
La afectación de las células presentadoras de antígeno
Pero aquí non acaba la cosa, tanto los factores virales antes descritos como los del huésped afectan la virulencia de las enfermedades por coronavirus en animales. Un ejemplo de ello es que tanto Covid-19 como SARS se caracterizan por una respuesta inflamatoria exacerbada y, de hecho, la carga viral no está correlacionada con el empeoramiento de síntomas. En este contexto es vital conocer el mecanismo que desencadena que la respuesta inflamatoria del hospedador, con sobreexpresiones de citocinas, se convierta en una causa principal de daño pulmonar y posterior mortalidad en los casos severos de Covid-19. Las dianas principales de la infección por los SARS-CoV son las células epiteliales ciliadas de la vía aérea y los pneumocitos alveolares de tipo II.
Pero también se ha demostrado que los SARS-CoV usan DC-SIGN y L-SIGN como correceptores para la entrada. Es de señalar que cada uno estos receptores son muy expresados en algunos tipos de células presentadoras de antígenos inmaduras (APC), sobre todo células dendríticas (DC) y podría aumentar el tropismo viral y por consiguiente contribuir aún más en la inmunopatogenia de la enfermedad. La alta infectividad viral en APC inmaduras indica que hay muchos PRR en ellas y que el virus puede superar la respuesta antiviral inicial. Pero también nos revela que la maduración de las APC no sólo conlleva una bajada dramática del número de PRR sino la aparición de otros mecanismos de restricción que evitan la replicación de virus. Estas y otras APC actúan como primera defensa contra la infección viral al estimular la vigilancia inmunológica, la defensa antiviral intrínseca y el puente entre la inmunidad innata y adaptativa participando en el reclutamiento de los linfocitos de memoria y de células efectoras al sitio de infección. Además, estas APC profesionales también son una fuente importante de interferones tipo I. Así, el efecto citopático del coronavirus es un mecanismo indirecto de sobrestimulación de APC y se asocia también con la inducción de inflamación y la secreción masiva de citocinas en los pulmones como IL-1, IL-6, IL-8, TNF-a y CXCL-10 que pueden provocar daños en los tejidos y favorecer la diseminación del virus sistémica.
Por otro lado, el mecanismo de escape inmune a través de la afectación de APCs resultaría en un aclaramiento viral ineficiente, lo que puede explicar la alta patogenicidad y las manifestaciones clínicas visto en Covid-19, MERS y SARS. Luego que las APC funcionen adecuadamente es crítico para la mitigar la infección y limitar el desarrollo de la enfermedad. En este sentido, existen tres posibles candidatos terapéuticos sobre la mesa, baricitinib, fedratinib y ruxolitinib que son inhibidores potentes y selectivos de JAK aprobados para indicaciones como artritis reumatoide y mielofibrosis Los tres son potentes antiinflamatorios que inhiben la señalización JAK-STAT de los receptores de citocinas. Desde el punto de vista técnico y de desarrollo de fármacos, esta permisividad de APC sobre coronavirus nos va a ayudar en el screening in vitro de potenciales agentes terapéuticos.
Consideraciones finales
En algunos individuos, la presencia del coronavirus en las vías respiratorias va a ser la puerta de entrada a otros microorganismos oportunistas que podrían ocasionar estas neumonías severas. Esta puerta se abre por el efecto citopático directo del virus sobre las células epiteliales del tracto respiratorio y por el daño tisular local que la respuesta inflamatoria va a ocasionar. Si a esto le añadimos un sistema inmunitario ineficiente debido a los mecanismos de inmunoevasión del coronavirus, tenemos todas las papeletas para convertir a Covid-19 en una enfermedad especialmente contagiosa, perdurable en el individuo y en algunos casos grave. Los mecanismos de escape viral e inmunopatogenia no son únicos de los coronavirus y han sido descritas muchas formas diferentes de interferir en la señalización de citocinas y de sus mecanismos de transducción. Además, otros virus de las familias de adenovirus, herpes, papiloma y poxvirus, entre otros interfieren en la actividad del proteasoma y el proceso celular dedicado a la presentación antigénica o de aparición del MHC en la membrana. Otros codifican para citocinas inmunosupresoras o cambios de fenotipos de los linfocitos helper (como el shift Th1 a Th2, poco eficiente para parásitos intracelulares). Generalmente son grandes virus de DNA y aquí los coronavirus representan una buena excepción a esta regla.
Hasta el momento, Covid-19 se está comportando como un enemigo de la especie humana, con unas tasas de letalidad difícilmente asumibles y que está poniendo en jaque el movimiento de personas, la globalización y la economía mundial. Sin ninguna duda, sus efectos sobre países en vías de desarrollo serán todavía peores. Por ello, como enemigo que es de todos nosotros, requiere un esfuerzo global para combatirlo. Destinemos los recursos necesarios para comprender mejor la patogenia, desarrollar antivirales y medidas de soporte más efectivas y luchemos por conseguir la vacuna preventiva. Tomemos las medidas oportunas para contener la diseminación de la enfermedad y protejamos a los más sensibles o susceptibles a sucumbir ante el virus, pero con cautela y sin alarmar innecesariamente a la población. Pero eso sí, no deberíamos permitir que Covid-19 se convierta en la pandemia del siglo XXI como sucedió con la gripe de 1918.
Referencias
Cong, Y., Hart, B. J., Gross, R., Zhou, H., Frieman, M., Bollinger, L., … Holbrook, M. R. (2018). MERS-CoV pathogenesis and antiviral efficacy of licensed drugs in human monocyte-derived antigen-presenting cells. PLoS ONE, 13(3), 1–17. https://doi.org/10.1371/journal.pone.0194868
Morales, D. J., & Lenschow, D. J. (2013). The antiviral activities of ISG15. Journal of Molecular Biology, 425(24), 4995–5008. https://doi.org/10.1016/j.jmb.2013.09.041
Cui, J., Li, F., & Shi, Z. L. (2019). Origin and evolution of pathogenic coronaviruses. Nature Reviews Microbiology, 17(3), 181–192. https://doi.org/10.1038/s41579-018-0118-9
Chen, Y., Liu, Q., & Guo, D. (2020). Emerging coronaviruses: Genome structure, replication, and pathogenesis. Journal of Medical Virology, (January), 418–423. https://doi.org/10.1002/jmv.25681
Stebbing, J., Phelan, A., Griffin, I., Tucker, C., Oechsle, O., Smith, D., & Richardson, P. (2020). COVID-19: combining antiviral and anti-inflammatory treatments. The Lancet Infectious Diseases, 2019(20), 2019–2020. https://doi.org/10.1016/S1473-3099(20)30132-8
Poulter, N. (2020). Lower Blood Pressure in South Asia? Trial Evidence. New England Journal of Medicine, 382(8), 758–760. https://doi.org/10.1056/NEJMe1917479
Zhu, N., Zhang, D., Wang, W., Li, X., Yang, B., Song, J., … Tan, W. (2020). A Novel Coronavirus from Patients with Pneumonia in China, 2019. New England Journal of Medicine, 727–733. https://doi.org/10.1056/nejmoa2001017
Totura, A. L., & Baric, R. S. (2012). SARS coronavirus pathogenesis: Host innate immune responses and viral antagonism of interferon. Current Opinion in Virology, 2(3), 264–275. https://doi.org/10.1016/j.coviro.2012.04.004
Li, F. (2013). Receptor recognition and cross-species infections of SARS coronavirus. Antiviral Research, 100(1), 246–254. https://doi.org/10.1016/j.antiviral.2013.08.014
Frieman, M., & Baric, R. (2008). Mechanisms of Severe Acute Respiratory Syndrome Pathogenesis and Innate Immunomodulation. Microbiology and Molecular Biology Reviews, 72(4), 672–685. https://doi.org/10.1128/mmbr.00015-08
Etiquetas: antiviral, citocina, coronavirus, Inmunopatogenia, interferon
← ¿Por qué un blog sobre inmunología?La tormenta de citocinas y su implicación en la patogenia del virus de la gripe A y del Covid-19 →
Funciona con WordPress / Tema Academica
Por Rafael Sirera en Inmunopatogenia sobre 09/03/2020
>CORONAVIRUS, INMUNOPATOGENIA VIRAL
.
los mecanismos que conducen a la inmunología y la patogenia del nuevo coronavirus SARS-CoV-2 son complejos
Un virus es capaz de penetrar a través de las barreras físicas que forma nuestro recubrimiento epitelial o mucoso, por ejemplo, en el tracto respiratorio. Y que es capaz de resistir las barreras químicas y biológicas creadas por nuestros microorganismos comensales, las células epiteliales y por las células inmunitarias que producen desagradables
ambientes ácidos, grasos y salinos o que secretan enzimas tóxicos o péptidos catiónicos que podrían destruir sus biomoléculas y con ello su integridad.
Si el virus ha sido capaz de saltarse esta barrera de elementos defensivos constitutivos y preformados ya lo tenemos en nuestro interior. Nos ha infectado. Ahora necesitamos que nuestras células sean capaces de percibirlo, que puedan iniciar la respuesta inmunitaria innata inducida y la inflamatoria con el objetivo de contenerlo y erradicarlo en menos de 96 horas. Pero en este contexto hay que tener en cuenta un detalle muy importante, el virus además de penetrar ha de dañar discretamente nuestros tejidos. Ello no debería ser ningún obstáculo para él, pues seguramente tendrá tropismo por alguna célula del epitelio, penetrará así en su interior, le secuestrará la maquinaria biosintética en su propio beneficio y generará nuevos viriones con el consiguiente estallido y necrosis de la célula infectada.
La detección de la infección
Con estas muertes celulares no programadas se van a liberan de forma masiva moléculas endógenas celulares que además van a dañar o modifica el andamiaje de los tejidos. Estas moléculas o cambios los denominamos patrones moleculares asociados al daño (también conocidos como DAMP o alarminas) y son inmunogénicos pues las células de la inmunidad innata poseen receptores que van a ser capaces de percibirlos y reconocerlos.
Pero no debemos olvidar que además del daño tenemos al extraño, al virus que ha conseguido infectarnos. Pues bien, los microorganismos patogénicos también tienen otros patrones moleculares asociados (en términos inmunológicos PAMP o MAMP) que serán reconocidos por los sensores celulares. Como los microorganismos pueden localizarse o esconderse en cualquier ubicación del individuo, sus células y especialmente las del sistema inmunitario, disponen de un gran abanico de sensores de estos patrones y los denominamos receptores que reconocen patrones (PRR). Estos numerosos PRRs pertenecen a distintas familias estructurales y así mismo cumplen funciones muy diferentes. Hay sensores que se localizan tanto en las membranas de la célula (por ejemplo, toll-like receptors o TLR, scavenger receptors o SR y los receptores de manosa de los macrófagos o MMR) como en el citoplasma de la misma (los NOD-like receptors o NLR y los RIG-like receptors o RLR), como en los humores o fluidos biológicos (la lectina que une manosa o MBL y la proteína C-reactiva o CRP). Este último tipo de PRR humorales los producen también otras células como los hepatocitos, que los liberan a la sangre para que se repartan por todo el organismo.
Estos PRRs además de la función como sensor cumplen otras dos funciones muy distintas y a la vez muy importantes para la defensa del individuo. Por un lado, la fagocitosis de los microorganismos que sean reconocidos por los neutrófilos y los macrófagos y, por otro lado, la activación del complemento, el principal ejecutor de la inmunidad innata humoral. Esta función tipo sensor va a disparar la respuesta inmunitaria innata local, la respuesta inflamatoria y la de reparación tisular. Se va a aumentar la permeabilidad de los vasos para que nuevos componentes del sistema inmunitario puedan acudir a este territorio que está siendo invadido por los virus y que la suma y la unión haga la fuerza antimicrobiana. También es muy destacable tener en cuenta que esta respuesta inflamatoria y la actividad de las células dendríticas y macrófagos presentando antígenos y estimulando a los linfocitos T, también va a llevar a la activación de la respuesta inmunitaria adaptativa. Esta última es la que en última instancia combatirá de la forma más específica, especializada y efectiva la infección del virus, este parásito intracelular y además podremos beneficiarnos de su efecto memoria ante futuras exposiciones del mismo microbio.
La respuesta antiviral intrínseca
Volvamos otra vez a fijarnos en el agente invasor, el virus, el cual debido a que es una partícula no viva y de absoluta simplicidad estructural podría ser muy resistente y esquivo a todas estas formas de reconocimiento y medidas que la respuesta inmunitaria esta generando contra el mismo. Pero nuestras células y nuestro sistema inmunitario han evolucionado a lo largo de millones de años de forma muy satisfactoria, conocedores de que existen los virus y que para poder detectarlos y contrarrestarlos son necesarias otras estrategias muy diferentes que las que generamos contra los otros tipos de microorganismos.
Yan N, Nat Immunol 2012
Y nos estamos refiriendo a la inmunidad antiviral intrínseca que restringe directamente la replicación viral y el ensamblaje, lo que hace que una célula no sea permisiva a la colonización viral. Esta inmunidad intrínseca es conferida por factores preexistentes y en gran medida por la presencia de factores inducidos por la propia infección del virus. Una de las formas más simples de impedir la actividad y replicación viral se observa en invertebrados y la denominamos inmunidad basada en el RNA al utiliza distintos tipos de RNA de interferencia que bloquearán la expresión de los genes virales e incluso inducirán su degradación. Este sistema es comparable a la regulación epigenética que realizan los microRNAs usando las enzimas Dicer y RISC o de una forma análoga al conocido sistema antiviral bacteriano CRISPR-Cas. Aumentando la complejidad evolutiva, los vertebrados hemos desarrollado otro mecanismo más complejo y eficiente, que es una inmunidad basada en proteínas y es la respuesta a los interferones (su nombre deriva de que interfiere en la capacidad infectiva de un virus). Como vamos a ver, la respuesta del interferón que desencadena alguno de los citados PRRs al detectar un virus no solo le afecta a la célula que percibe este invasor (autocrina), sino que se extenderá de forma paracrina a las células de su vecindario para que todas puedan degradar la información genética del virus e impidan la expresión de sus genes.
La respuesta del interferón
Llegado este momento es importante que veamos de forma algo más detallado en qué consiste la respuesta antiviral mediada por interferón. Los principales PRR que detectan la presencia de virus se basan en localizar ácidos nucleicos no “naturales” de la célula como dsRNA y DNA con CpG no metilados, y se localizan en las membranas de las vesículas endocíticas (dentro de la célula) y son los TLR3, 7, 8 y 9, además de los RLR citosólicos RIG-I y MDA5. Mediante un mecanismo complejo de señalización y transducción estos PRR consiguen que se fosforilen y dimericen componentes de una familia de factores de transcripción que se denominan IRF (interferon regulatory factor) y que puedan translocarse al núcleo. Una vez en el núcleo, estos IRF interaccionaran con genes que contienen en sus promotores secuencias nucleotídicas denominadas ISRE (IFN-stimulated response elements) y que conducen a la expresión de los interferones de tipo I (INF-a e IFN-b) y otros genes antivirales importantes (que ahora luego volveremos con ellos).
Gilliet M, Nat Rev immune 2008
Hay que hacer notar que este interferón que va a producir la célula no interfiere directamente con el virus, sino que va a ser secretado y las células que posean receptores para los interferones de tipo I (IFNAR) serán las que se beneficien de su producción y liberación. Afortunadamente la expresión del receptor de IFN se transcribe en ese contexto de ISRE por lo que la célula productora será una de las beneficiadas junto con otras células del microambiente. La señalización a través de los IFNAR lleva a un mecanismo similar donde intervienen otros factores de transcripción que actúan sobre las mismas secuencias ISRE y alimentan como un bucle con feedback positivo todo el proceso de señalización y protección local contra los virus.
Michalska A, Front Immunol 2018
Nos queda ver ahora cuales esas proteínas que se van a producir gracias a los interferones y son la que directamente van en contra de los virus. En primer lugar, se aumenta la expresión del complejo principal de histocompatibilidad de tipo I y la presentación de proteínas celulares (y también virales, pues el virus está dentro de la célula y usa su maquinaria metabólica) a los linfocitos T. Este proceso fundamental para la vigilancia inmunológica permite que los linfocitos detecten la producción de proteínas extrañas al organismo (como las virales) y si se da el caso, que se genere la respuesta inmunitaria adaptativa. Todo ello acompañado en un contexto que maximiza la activación de los linfocitos citotóxicos (CTL) que en última instancia serán los encargados de destruir a células infectadas por virus. Además de la activación de los CTL, la célula produce una serie de enzimas que degradan RNAs virales como la 2′,5′ oligoadenilato sintetasa, PKR que impiden la acción de factores de transcripción necesarios para la expresión de proteínas virales y proteasas que degradan las proteínas de la cápside viral que se hayan sintetizado, impidiendo el ensamblaje de nuevos viriones. De esta forma tan eficiente la inmunidad innata logra detectar los virus y no solo luchar contra ellos, sino que iniciar la activación de la respuesta inmunitaria adaptativa.
Abbas A, Cellular and Molecular Immunology, Elsevier
Pero los virus no iban a ser menos que las células de los vertebrados y su proceso evolutivo a algunos también les ha permitido adquirir mecanismos de resistencia o de evasión de la respuesta antiviral y por ende de la respuesta inmunitaria contribuyendo a la inmunopatogenia de algunas enfermedades virales. Estas estrategias son muy variadas y están asociadas a la complejidad genómica del virus. Cuantos más genes sean capaces de empaquetar en sus cápsides, seguramente más genes tendrán para bloquear la señalización de PRR y de IFNAR. Para poder entenderlo mejor, lo más conveniente será ver un ejemplo y en estos momentos tal vez el ejemplo más interesante es el coronavirus. Vayamos pues a ver los mecanismos inmunopatogénicos de los coronavirus
Un ejemplo concreto, el Coronavirus y la epidemia Covid-19
En diciembre del año 2019 la humanidad ha sufrido por tercera vez en las últimas dos décadas una nueva zoonosis causada por un coronavirus (SARS-CoV-2), provocando una epidemia que afecta a las vías respiratorias y que en algunos caos es una enfermedad muy grave, la denominada Covid-19. El avance la biología molecular y el riesgo a que se convierta en una pandemia ha intensificado los esfuerzos de los centros de investigación, las autoridades sanitarias de muchos países y de la OMS. Gracias a ello, en tan poco tiempo tenemos muchos datos epidemiológicos, conocemos la información genética del virus y se han desarrollado test diagnósticos muy sensibles y cuantitativos. Se ha podido establecer el tropismo viral sobre células epiteliales alveolares gracias a la interacción con la exopeptidasa, enzima convertidora de angiotensina (hACE2). Esta enzima transforma, preferentemente en el pulmón, al precursor de la angiotensina en una molécula activa y que se encarga de regular la presión arterial. Pero aún quedan muchas lagunas por resolver y que nos pueden ayudar a contener la epidemia, como cuál ha sido el vector, el desarrollo de algún antiviral o de una vacuna. A pesar de todo, el esfuerzo tecnológico y humano ha sido tremendo y sin parangón.
Cui J, Nat Rev Microbiol 2019
El virus y sus genes estructurales
Los Coronavirus son virus de RNA monocatenario con capacidad codificante (+ssRNA) y en este caso concreto, el tamaño del genoma de SARS-CoV-2 es de 30 kb. Teniendo en cuenta que 10 kb es el tamaño medio de los virus de RNA, este detalle sitúa a los coronavirus como los virus de RNA con genoma mas grande.
Banerjee A, Curr Clin Micro Rpt (2019)
Y tener un genoma mayor conlleva mejoras en su supervivencia y mejoras en su adaptación al hospedador lo que implica en muchos casos capacidad de escape inmunológico o cierta inmunopatogenia. Esta complejidad, en primer lugar, se asocia con una característica muy relevante a la hora de la multiplicación y la evolución viral y es que el complejo de replicación transcripción tiene actividad 3’‐5’ exorribonucleasa, lo que le confiere mayor fidelidad de copia. Luego los coronavirus mutan, seguro que mucho, pero menos de lo que cabría esperar tiendo en cuenta que es un virus de RNA. Las proteínas que están codificadas en su genoma se dividen en dos grupos, proteínas no estructurales y proteínas estructurales.
Chen Y, J Med Virol 2020
Empecemos por estas últimas, y dentro de las estructurales, en sentido 5’-3’ tenemos:
La proteína S (Spike, pincho), que formando homotrímeros crea esas estructuras que asoman sobre la nuceocápside y le permiten anclarse a su receptor (hACE2).
La proteína E (Envuelta) encargada del ensamblaje viral y la patogenia del virus.
La proteína M (Membrana) tiene tres dominios transmembrana y al unirse a la nucleocápside le confiere la estructura esférica.
La proteína N (Nucleocápside) tiene dos dominios, uno que interacciona con el RNA viral conduciendo a su correcto encapsulamiento en los viriones y otro dominio multifunción que actúa por un lado como antagonista del IFN y por otro lado como represor de los RNA de interferencia, lo cual es muy importante para evitar la acción inmunitaria antiviral y es beneficios para su replicación.
Además, los b-Coronavirus también disponen de la hemaglutinina-acetilesterasa (HE), una glicoproteína que se une a restos de azúcar en las membranas celulares e induce la fusión y penetración viral en la célula. Curiosamente, parece ser que el gen para HE fue introducido en un genoma ancestral de coronavirus por recombinación con el virus de la influenza C.
Holmes KV, N Engl J Med 2003
Los genes no estructurales y la inmunopatogenia viral
El análisis genético y molecular del SARS-CoV-2 nos permite afirmar que no es ni un mutante de cualquier coronavirus anterior ni un recombinante de coronavirus conocidos. Es un coronavirus desconocido, probablemente de un hospedador no humano que de alguna manera adquirió la capacidad de infectar a humanos y provocar la presente zoonosis.
Zhu N, N Engl J Med 2020
Sobre los genes no estructurales empezaremos abordando el papel que juegan las otras proteínas virales, así:
La nsp13 es una helicasa, una enzima vital para el desempaquetado de los genes y por ende la replicación y transcripción.
Las nsp7 a 10 son regiones críticas como reguladoras de la nsp12, que es la RNA polimerasa multidominio y que junto con la actividad de reparación del RNA (MMR) de la nsp14 le confiere estas altas tasas de fidelidad de copia.
Por otro lado, como la síntesis del mRNA del coronavirus se realiza en el citosol en lugar del núcleo, los virus de +RNA de eucariotas no pueden confiar en que las proteínas nucleares de la célula se encarguen de añadir la 7-metilguanosina (m7G) al primer nucleótido del RNA para protegerlo de las exorribonucleasas celulares (proceso que se denomina capping) y conducir a su traducción en proteínas. En este sentido, las nsp10-13-14-16 virales participan en el capping del RNA viral.
Veamos entonces las claves de la inmunopatogenia de los coronavirus que pivotan sobre el bloque de la actividad antiviral, que se pone de manifiesto gracias a la acción del resto de proteínas no estructurales:
La nsp1 de los SARS-CoV es una proteína de 20 kDa que se localiza en el citoplasma de las células infectadas y que genera una variedad de funciones únicas en los b-Coronavirus. En primer lugar, es capaz de bloquear y degradar el mRNA del IFN-b y otros mensajeros endógenos. En segundo lugar, al bloquear la fosforilación de STAT1 e I-kB y la dimerización de IRF3, inhibe las vías de transducción de señales que involucran IRF, STAT y NF-k Ello afecta también a la progresión del ciclo celular sin inducir apoptosis, favoreciendo la supervivencia viral.
La nsp3, codificada en el gran ORF1 y que genera una proteína de 200 kDa con varios dominios funcionales diferentes. Estos incluyen un dominio de unión de poli (ADP-ribosa), de proteasa (papain-like protease, PLP), otro deubiquitinasa y un último de ISGylase. La función PLP de nsp3 es la que antagoniza las respuestas IFN de tipo I, impidiendo la fosforilación, dimerización e importe al núcleo de IRF3. De forma análoga actúa sobre la vía de represión de NF-k Además, la función ISGylase contrarresta la acción del gen 15 estimulado por interferón (ISG15), que es la ISGilación. Veamos en qué consiste este proceso. La modificación postraduccional de las proteínas es una estrategia importante para la regulación del proteoma celular. La ubiquitina y otros modificadores similares a la ubiquitina como ISG15, a través de su conjugación covalente a proteínas median la regulación de los niveles de esas proteínas, las vías de señalización, el tráfico vesicular y muchos otros procesos celulares. ISG15 se codifica y se expresa como una proteína precursora de 17 kDa que se procesa proteolíticamente en su extremo C-ter para exponer una secuencia de aminoácidos, idéntica a la secuencia C-ter de la ubiquitina, y sus adiciones covalentes a las proteínas son bastante similares. Por este mecanismo se regula a la baja la vida media de las proteínas virales, disminuyendo su capacidad de ensamblaje y la viabilidad de los mismos. De forma contraria este proceso afecta aumentando la vida media de muchos genes regulados al alza como respuesta de IRF. Por este motivo esta actividad ISGylase parece una estrategia acertada de propagación viral.
Morales DJ, J Mol Biol 2013
Las otras nsp4 a nsp6 bloquean directamente la actividad antiviral que la célula debería poner en marcha inhibiendo la señalización del IFN. Algunas formas descritas por SARS-CoVs de antagonizar a los IFN de tipo I es mediante la inactivación de la traducción del hospedador; la degradación de sus mARN; interactuar negativamente con STING; antagonizar la señalización de IFN inducida por MAVS/RIG-I. Así mismo, también lo consiguen bloqueando la expresión de citocinas proinflamatorias y la generación de las vesículas que son necesarias para la formación de autofagosomas y el incremento de la presentación de péptidos virales.
Por último, la nsp16 es una 2’O metiltransferasa (2’‐O‐MTasa) que bloquea muy selectivamente la señalización a través de MDA5, un tipo de RIG-like receptor.
Tortura AL, Curr Opinion Virol 2012
La afectación de las células presentadoras de antígeno
Pero aquí non acaba la cosa, tanto los factores virales antes descritos como los del huésped afectan la virulencia de las enfermedades por coronavirus en animales. Un ejemplo de ello es que tanto Covid-19 como SARS se caracterizan por una respuesta inflamatoria exacerbada y, de hecho, la carga viral no está correlacionada con el empeoramiento de síntomas. En este contexto es vital conocer el mecanismo que desencadena que la respuesta inflamatoria del hospedador, con sobreexpresiones de citocinas, se convierta en una causa principal de daño pulmonar y posterior mortalidad en los casos severos de Covid-19. Las dianas principales de la infección por los SARS-CoV son las células epiteliales ciliadas de la vía aérea y los pneumocitos alveolares de tipo II.
Pero también se ha demostrado que los SARS-CoV usan DC-SIGN y L-SIGN como correceptores para la entrada. Es de señalar que cada uno estos receptores son muy expresados en algunos tipos de células presentadoras de antígenos inmaduras (APC), sobre todo células dendríticas (DC) y podría aumentar el tropismo viral y por consiguiente contribuir aún más en la inmunopatogenia de la enfermedad. La alta infectividad viral en APC inmaduras indica que hay muchos PRR en ellas y que el virus puede superar la respuesta antiviral inicial. Pero también nos revela que la maduración de las APC no sólo conlleva una bajada dramática del número de PRR sino la aparición de otros mecanismos de restricción que evitan la replicación de virus. Estas y otras APC actúan como primera defensa contra la infección viral al estimular la vigilancia inmunológica, la defensa antiviral intrínseca y el puente entre la inmunidad innata y adaptativa participando en el reclutamiento de los linfocitos de memoria y de células efectoras al sitio de infección. Además, estas APC profesionales también son una fuente importante de interferones tipo I. Así, el efecto citopático del coronavirus es un mecanismo indirecto de sobrestimulación de APC y se asocia también con la inducción de inflamación y la secreción masiva de citocinas en los pulmones como IL-1, IL-6, IL-8, TNF-a y CXCL-10 que pueden provocar daños en los tejidos y favorecer la diseminación del virus sistémica.
Por otro lado, el mecanismo de escape inmune a través de la afectación de APCs resultaría en un aclaramiento viral ineficiente, lo que puede explicar la alta patogenicidad y las manifestaciones clínicas visto en Covid-19, MERS y SARS. Luego que las APC funcionen adecuadamente es crítico para la mitigar la infección y limitar el desarrollo de la enfermedad. En este sentido, existen tres posibles candidatos terapéuticos sobre la mesa, baricitinib, fedratinib y ruxolitinib que son inhibidores potentes y selectivos de JAK aprobados para indicaciones como artritis reumatoide y mielofibrosis Los tres son potentes antiinflamatorios que inhiben la señalización JAK-STAT de los receptores de citocinas. Desde el punto de vista técnico y de desarrollo de fármacos, esta permisividad de APC sobre coronavirus nos va a ayudar en el screening in vitro de potenciales agentes terapéuticos.
Consideraciones finales
En algunos individuos, la presencia del coronavirus en las vías respiratorias va a ser la puerta de entrada a otros microorganismos oportunistas que podrían ocasionar estas neumonías severas. Esta puerta se abre por el efecto citopático directo del virus sobre las células epiteliales del tracto respiratorio y por el daño tisular local que la respuesta inflamatoria va a ocasionar. Si a esto le añadimos un sistema inmunitario ineficiente debido a los mecanismos de inmunoevasión del coronavirus, tenemos todas las papeletas para convertir a Covid-19 en una enfermedad especialmente contagiosa, perdurable en el individuo y en algunos casos grave. Los mecanismos de escape viral e inmunopatogenia no son únicos de los coronavirus y han sido descritas muchas formas diferentes de interferir en la señalización de citocinas y de sus mecanismos de transducción. Además, otros virus de las familias de adenovirus, herpes, papiloma y poxvirus, entre otros interfieren en la actividad del proteasoma y el proceso celular dedicado a la presentación antigénica o de aparición del MHC en la membrana. Otros codifican para citocinas inmunosupresoras o cambios de fenotipos de los linfocitos helper (como el shift Th1 a Th2, poco eficiente para parásitos intracelulares). Generalmente son grandes virus de DNA y aquí los coronavirus representan una buena excepción a esta regla.
Hasta el momento, Covid-19 se está comportando como un enemigo de la especie humana, con unas tasas de letalidad difícilmente asumibles y que está poniendo en jaque el movimiento de personas, la globalización y la economía mundial. Sin ninguna duda, sus efectos sobre países en vías de desarrollo serán todavía peores. Por ello, como enemigo que es de todos nosotros, requiere un esfuerzo global para combatirlo. Destinemos los recursos necesarios para comprender mejor la patogenia, desarrollar antivirales y medidas de soporte más efectivas y luchemos por conseguir la vacuna preventiva. Tomemos las medidas oportunas para contener la diseminación de la enfermedad y protejamos a los más sensibles o susceptibles a sucumbir ante el virus, pero con cautela y sin alarmar innecesariamente a la población. Pero eso sí, no deberíamos permitir que Covid-19 se convierta en la pandemia del siglo XXI como sucedió con la gripe de 1918.
Referencias
Cong, Y., Hart, B. J., Gross, R., Zhou, H., Frieman, M., Bollinger, L., … Holbrook, M. R. (2018). MERS-CoV pathogenesis and antiviral efficacy of licensed drugs in human monocyte-derived antigen-presenting cells. PLoS ONE, 13(3), 1–17. https://doi.org/10.1371/journal.pone.0194868
Morales, D. J., & Lenschow, D. J. (2013). The antiviral activities of ISG15. Journal of Molecular Biology, 425(24), 4995–5008. https://doi.org/10.1016/j.jmb.2013.09.041
Cui, J., Li, F., & Shi, Z. L. (2019). Origin and evolution of pathogenic coronaviruses. Nature Reviews Microbiology, 17(3), 181–192. https://doi.org/10.1038/s41579-018-0118-9
Chen, Y., Liu, Q., & Guo, D. (2020). Emerging coronaviruses: Genome structure, replication, and pathogenesis. Journal of Medical Virology, (January), 418–423. https://doi.org/10.1002/jmv.25681
Stebbing, J., Phelan, A., Griffin, I., Tucker, C., Oechsle, O., Smith, D., & Richardson, P. (2020). COVID-19: combining antiviral and anti-inflammatory treatments. The Lancet Infectious Diseases, 2019(20), 2019–2020. https://doi.org/10.1016/S1473-3099(20)30132-8
Poulter, N. (2020). Lower Blood Pressure in South Asia? Trial Evidence. New England Journal of Medicine, 382(8), 758–760. https://doi.org/10.1056/NEJMe1917479
Zhu, N., Zhang, D., Wang, W., Li, X., Yang, B., Song, J., … Tan, W. (2020). A Novel Coronavirus from Patients with Pneumonia in China, 2019. New England Journal of Medicine, 727–733. https://doi.org/10.1056/nejmoa2001017
Totura, A. L., & Baric, R. S. (2012). SARS coronavirus pathogenesis: Host innate immune responses and viral antagonism of interferon. Current Opinion in Virology, 2(3), 264–275. https://doi.org/10.1016/j.coviro.2012.04.004
Li, F. (2013). Receptor recognition and cross-species infections of SARS coronavirus. Antiviral Research, 100(1), 246–254. https://doi.org/10.1016/j.antiviral.2013.08.014
Frieman, M., & Baric, R. (2008). Mechanisms of Severe Acute Respiratory Syndrome Pathogenesis and Innate Immunomodulation. Microbiology and Molecular Biology Reviews, 72(4), 672–685. https://doi.org/10.1128/mmbr.00015-08
Etiquetas: antiviral, citocina, coronavirus, Inmunopatogenia, interferon
← ¿Por qué un blog sobre inmunología?La tormenta de citocinas y su implicación en la patogenia del virus de la gripe A y del Covid-19 →
Funciona con WordPress / Tema Academica
Por Rafael Sirera en Inmunopatogenia sobre 09/03/2020