 >CORONAVIRUS, INMUNOPATOGENIA VIRAL
>CORONAVIRUS, INMUNOPATOGENIA VIRAL
.
los mecanismos que conducen a la inmunología y la patogenia del nuevo coronavirus SARS-CoV-2 son complejos
Un virus es capaz de penetrar a través de las barreras físicas que forma nuestro recubrimiento epitelial o mucoso, por ejemplo, en el tracto respiratorio. Y que es capaz de resistir las barreras químicas y biológicas creadas por nuestros microorganismos comensales, las células epiteliales y por las células inmunitarias que producen desagradables
ambientes ácidos, grasos y salinos o que secretan enzimas tóxicos o péptidos catiónicos que podrían destruir sus biomoléculas y con ello su integridad.
Si el virus ha sido capaz de saltarse esta barrera de elementos defensivos constitutivos y preformados ya lo tenemos en nuestro interior. Nos ha infectado. Ahora necesitamos que nuestras células sean capaces de percibirlo, que puedan iniciar la respuesta inmunitaria innata inducida y la inflamatoria con el objetivo de contenerlo y erradicarlo en menos de 96 horas. Pero en este contexto hay que tener en cuenta un detalle muy importante, el virus además de penetrar ha de dañar discretamente nuestros tejidos. Ello no debería ser ningún obstáculo para él, pues seguramente tendrá tropismo por alguna célula del epitelio, penetrará así en su interior, le secuestrará la maquinaria biosintética en su propio beneficio y generará nuevos viriones con el consiguiente estallido y necrosis de la célula infectada.
La detección de la infección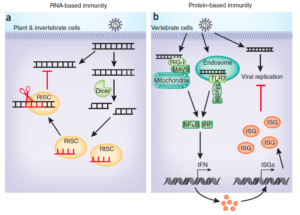
Con estas muertes celulares no programadas se van a liberan de forma masiva moléculas endógenas celulares que además van a dañar o modifica el andamiaje de los tejidos. Estas moléculas o cambios los denominamos patrones moleculares asociados al daño (también conocidos como DAMP o alarminas) y son inmunogénicos pues las células de la inmunidad innata poseen receptores que van a ser capaces de percibirlos y reconocerlos.
Pero no debemos olvidar que además del daño tenemos al extraño, al virus que ha conseguido infectarnos. Pues bien, los microorganismos patogénicos también tienen otros patrones moleculares asociados (en términos inmunológicos PAMP o MAMP) que serán reconocidos por los sensores celulares. Como los microorganismos pueden localizarse o esconderse en cualquier ubicación del individuo, sus células y especialmente las del sistema inmunitario, disponen de un gran abanico de sensores de estos patrones y los denominamos receptores que reconocen patrones (PRR). Estos numerosos PRRs pertenecen a distintas familias estructurales y así mismo cumplen funciones muy diferentes. Hay sensores que se localizan tanto en las membranas de la célula (por ejemplo, toll-like receptors o TLR, scavenger receptors o SR y los receptores de manosa de los macrófagos o MMR) como en el citoplasma de la misma (los NOD-like receptors o NLR y los RIG-like receptors o RLR), como en los humores o fluidos biológicos (la lectina que une manosa o MBL y la proteína C-reactiva o CRP). Este último tipo de PRR humorales los producen también otras células como los hepatocitos, que los liberan a la sangre para que se repartan por todo el organismo.
Estos PRRs además de la función como sensor cumplen otras dos funciones muy distintas y a la vez muy importantes para la defensa del individuo. Por un lado, la fagocitosis de los microorganismos que sean reconocidos por los neutrófilos y los macrófagos y, por otro lado, la activación del complemento, el principal ejecutor de la inmunidad innata humoral. Esta función tipo sensor va a disparar la respuesta inmunitaria innata local, la respuesta inflamatoria y la de reparación tisular. Se va a aumentar la permeabilidad de los vasos para que nuevos componentes del sistema inmunitario puedan acudir a este territorio que está siendo invadido por los virus y que la suma y la unión haga la fuerza antimicrobiana. También es muy destacable tener en cuenta que esta respuesta inflamatoria y la actividad de las células dendríticas y macrófagos presentando antígenos y estimulando a los linfocitos T, también va a llevar a la activación de la respuesta inmunitaria adaptativa. Esta última es la que en última instancia combatirá de la forma más específica, especializada y efectiva la infección del virus, este parásito intracelular y además podremos beneficiarnos de su efecto memoria ante futuras exposiciones del mismo microbio.
La respuesta antiviral intrínseca
Volvamos otra vez a fijarnos en el agente invasor, el virus, el cual debido a que es una partícula no viva y de absoluta simplicidad estructural podría ser muy resistente y esquivo a todas estas formas de reconocimiento y medidas que la respuesta inmunitaria esta generando contra el mismo. Pero nuestras células y nuestro sistema inmunitario han evolucionado a lo largo de millones de años de forma muy satisfactoria, conocedores de que existen los virus y que para poder detectarlos y contrarrestarlos son necesarias otras estrategias muy diferentes que las que generamos contra los otros tipos de microorganismos.
Yan N, Nat Immunol 2012
Y nos estamos refiriendo a la inmunidad antiviral intrínseca que restringe directamente la replicación viral y el ensamblaje, lo que hace que una célula no sea permisiva a la colonización viral. Esta inmunidad intrínseca es conferida por factores preexistentes y en gran medida por la presencia de factores inducidos por la propia infección del virus. Una de las formas más simples de impedir la actividad y replicación viral se observa en invertebrados y la denominamos inmunidad basada en el RNA al utiliza distintos tipos de RNA de interferencia que bloquearán la expresión de los genes virales e incluso inducirán su degradación. Este sistema es comparable a la regulación epigenética que realizan los microRNAs usando las enzimas Dicer y RISC o de una forma análoga al conocido sistema antiviral bacteriano CRISPR-Cas. Aumentando la complejidad evolutiva, los vertebrados hemos desarrollado otro mecanismo más complejo y eficiente, que es una inmunidad basada en proteínas y es la respuesta a los interferones (su nombre deriva de que interfiere en la capacidad infectiva de un virus). Como vamos a ver, la respuesta del interferón que desencadena alguno de los citados PRRs al detectar un virus no solo le afecta a la célula que percibe este invasor (autocrina), sino que se extenderá de forma paracrina a las células de su vecindario para que todas puedan degradar la información genética del virus e impidan la expresión de sus genes.
La respuesta del interferón
Llegado este momento es importante que veamos de forma algo más detallado en qué consiste la respuesta antiviral mediada por interferón. Los principales PRR que detectan la presencia de virus se basan en localizar ácidos nucleicos no “naturales” de la célula como dsRNA y DNA con CpG no metilados, y se localizan en las membranas de las vesículas endocíticas (dentro de la célula) y son los TLR3, 7, 8 y 9, además de los RLR citosólicos RIG-I y MDA5. Mediante un mecanismo complejo de señalización y transducción estos PRR consiguen que se fosforilen y dimericen componentes de una familia de factores de transcripción que se denominan IRF (interferon regulatory factor) y que puedan translocarse al núcleo. Una vez en el núcleo, estos IRF interaccionaran con genes que contienen en sus promotores secuencias nucleotídicas denominadas ISRE (IFN-stimulated response elements) y que conducen a la expresión de los interferones de tipo I (INF-a e IFN-b) y otros genes antivirales importantes (que ahora luego volveremos con ellos).
Gilliet M, Nat Rev immune 2008
Hay que hacer notar que este interferón que va a producir la célula no interfiere directamente con el virus, sino que va a ser secretado y las células que posean receptores para los interferones de tipo I (IFNAR) serán las que se beneficien de su producción y liberación. Afortunadamente la expresión del receptor de IFN se transcribe en ese contexto de ISRE por lo que la célula productora será una de las beneficiadas junto con otras células del microambiente. La señalización a través de los IFNAR lleva a un mecanismo similar donde intervienen otros factores de transcripción que actúan sobre las mismas secuencias ISRE y alimentan como un bucle con feedback positivo todo el proceso de señalización y protección local contra los virus.
Michalska A, Front Immunol 2018
Nos queda ver ahora cuales esas proteínas que se van a producir gracias a los interferones y son la que directamente van en contra de los virus. En primer lugar, se aumenta la expresión del complejo principal de histocompatibilidad de tipo I y la presentación de proteínas celulares (y también virales, pues el virus está dentro de la célula y usa su maquinaria metabólica) a los linfocitos T. Este proceso fundamental para la vigilancia inmunológica permite que los linfocitos detecten la producción de proteínas extrañas al organismo (como las virales) y si se da el caso, que se genere la respuesta inmunitaria adaptativa. Todo ello acompañado en un contexto que maximiza la activación de los linfocitos citotóxicos (CTL) que en última instancia serán los encargados de destruir a células infectadas por virus. Además de la activación de los CTL, la célula produce una serie de enzimas que degradan RNAs virales como la 2′,5′ oligoadenilato sintetasa, PKR que impiden la acción de factores de transcripción necesarios para la expresión de proteínas virales y proteasas que degradan las proteínas de la cápside viral que se hayan sintetizado, impidiendo el ensamblaje de nuevos viriones. De esta forma tan eficiente la inmunidad innata logra detectar los virus y no solo luchar contra ellos, sino que iniciar la activación de la respuesta inmunitaria adaptativa.
Abbas A, Cellular and Molecular Immunology, Elsevier
Pero los virus no iban a ser menos que las células de los vertebrados y su proceso evolutivo a algunos también les ha permitido adquirir mecanismos de resistencia o de evasión de la respuesta antiviral y por ende de la respuesta inmunitaria contribuyendo a la inmunopatogenia de algunas enfermedades virales. Estas estrategias son muy variadas y están asociadas a la complejidad genómica del virus. Cuantos más genes sean capaces de empaquetar en sus cápsides, seguramente más genes tendrán para bloquear la señalización de PRR y de IFNAR. Para poder entenderlo mejor, lo más conveniente será ver un ejemplo y en estos momentos tal vez el ejemplo más interesante es el coronavirus. Vayamos pues a ver los mecanismos inmunopatogénicos de los coronavirus
Un ejemplo concreto, el Coronavirus y la epidemia Covid-19
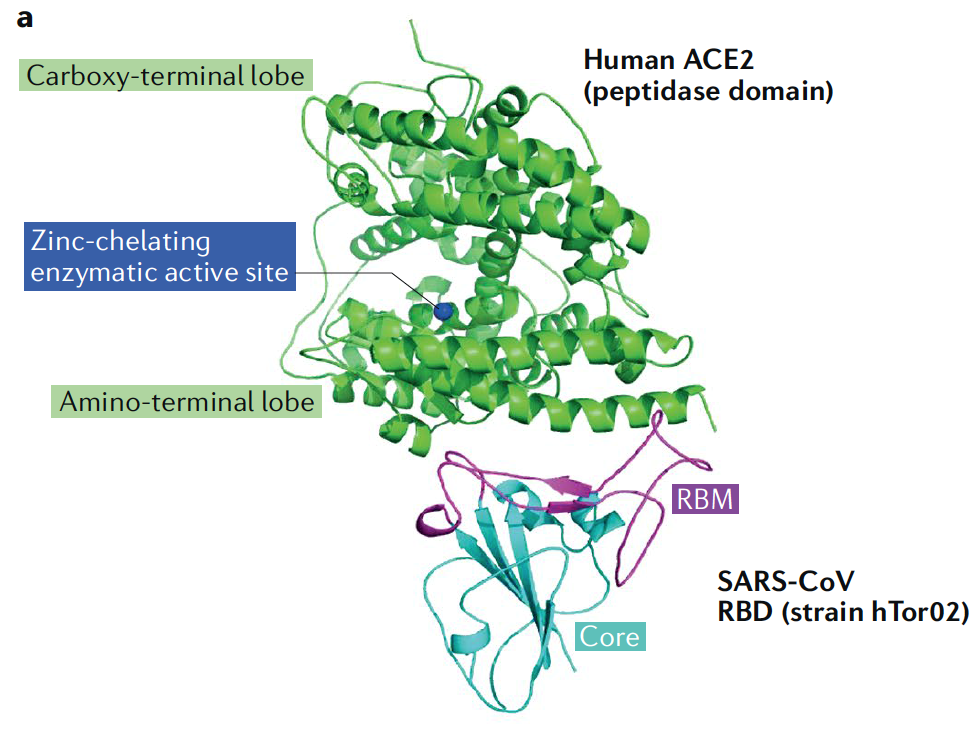
En diciembre del año 2019 la humanidad ha sufrido por tercera vez en las últimas dos décadas una nueva zoonosis causada por un coronavirus (SARS-CoV-2), provocando una epidemia que afecta a las vías respiratorias y que en algunos caos es una enfermedad muy grave, la denominada Covid-19. El avance la biología molecular y el riesgo a que se convierta en una pandemia ha intensificado los esfuerzos de los centros de investigación, las autoridades sanitarias de muchos países y de la OMS. Gracias a ello, en tan poco tiempo tenemos muchos datos epidemiológicos, conocemos la información genética del virus y se han desarrollado test diagnósticos muy sensibles y cuantitativos. Se ha podido establecer el tropismo viral sobre células epiteliales alveolares gracias a la interacción con la exopeptidasa, enzima convertidora de angiotensina (hACE2). Esta enzima transforma, preferentemente en el pulmón, al precursor de la angiotensina en una molécula activa y que se encarga de regular la presión arterial. Pero aún quedan muchas lagunas por resolver y que nos pueden ayudar a contener la epidemia, como cuál ha sido el vector, el desarrollo de algún antiviral o de una vacuna. A pesar de todo, el esfuerzo tecnológico y humano ha sido tremendo y sin parangón.
Cui J, Nat Rev Microbiol 2019
El virus y sus genes estructurales
Los Coronavirus son virus de RNA monocatenario con capacidad codificante (+ssRNA) y en este caso concreto, el tamaño del genoma de SARS-CoV-2 es de 30 kb. Teniendo en cuenta que 10 kb es el tamaño medio de los virus de RNA, este detalle sitúa a los coronavirus como los virus de RNA con genoma mas grande.
Banerjee A, Curr Clin Micro Rpt (2019)
Y tener un genoma mayor conlleva mejoras en su supervivencia y mejoras en su adaptación al hospedador lo que implica en muchos casos capacidad de escape inmunológico o cierta inmunopatogenia. Esta complejidad, en primer lugar, se asocia con una característica muy relevante a la hora de la multiplicación y la evolución viral y es que el complejo de replicación transcripción tiene actividad 3’‐5’ exorribonucleasa, lo que le confiere mayor fidelidad de copia. Luego los coronavirus mutan, seguro que mucho, pero menos de lo que cabría esperar tiendo en cuenta que es un virus de RNA. Las proteínas que están codificadas en su genoma se dividen en dos grupos, proteínas no estructurales y proteínas estructurales.
Chen Y, J Med Virol 2020
Empecemos por estas últimas, y dentro de las estructurales, en sentido 5’-3’ tenemos:
La proteína S (Spike, pincho), que formando homotrímeros crea esas estructuras que asoman sobre la nuceocápside y le permiten anclarse a su receptor (hACE2).
La proteína E (Envuelta) encargada del ensamblaje viral y la patogenia del virus.
La proteína M (Membrana) tiene tres dominios transmembrana y al unirse a la nucleocápside le confiere la estructura esférica.
La proteína N (Nucleocápside) tiene dos dominios, uno que interacciona con el RNA viral conduciendo a su correcto encapsulamiento en los viriones y otro dominio multifunción que actúa por un lado como antagonista del IFN y por otro lado como represor de los RNA de interferencia, lo cual es muy importante para evitar la acción inmunitaria antiviral y es beneficios para su replicación.
Además, los b-Coronavirus también disponen de la hemaglutinina-acetilesterasa (HE), una glicoproteína que se une a restos de azúcar en las membranas celulares e induce la fusión y penetración viral en la célula. Curiosamente, parece ser que el gen para HE fue introducido en un genoma ancestral de coronavirus por recombinación con el virus de la influenza C.
Holmes KV, N Engl J Med 2003
Los genes no estructurales y la inmunopatogenia viral
El análisis genético y molecular del SARS-CoV-2 nos permite afirmar que no es ni un mutante de cualquier coronavirus anterior ni un recombinante de coronavirus conocidos. Es un coronavirus desconocido, probablemente de un hospedador no humano que de alguna manera adquirió la capacidad de infectar a humanos y provocar la presente zoonosis.
Zhu N, N Engl J Med 2020
Sobre los genes no estructurales empezaremos abordando el papel que juegan las otras proteínas virales, así:
La nsp13 es una helicasa, una enzima vital para el desempaquetado de los genes y por ende la replicación y transcripción.
Las nsp7 a 10 son regiones críticas como reguladoras de la nsp12, que es la RNA polimerasa multidominio y que junto con la actividad de reparación del RNA (MMR) de la nsp14 le confiere estas altas tasas de fidelidad de copia.
Por otro lado, como la síntesis del mRNA del coronavirus se realiza en el citosol en lugar del núcleo, los virus de +RNA de eucariotas no pueden confiar en que las proteínas nucleares de la célula se encarguen de añadir la 7-metilguanosina (m7G) al primer nucleótido del RNA para protegerlo de las exorribonucleasas celulares (proceso que se denomina capping) y conducir a su traducción en proteínas. En este sentido, las nsp10-13-14-16 virales participan en el capping del RNA viral.
Veamos entonces las claves de la inmunopatogenia de los coronavirus que pivotan sobre el bloque de la actividad antiviral, que se pone de manifiesto gracias a la acción del resto de proteínas no estructurales:
La nsp1 de los SARS-CoV es una proteína de 20 kDa que se localiza en el citoplasma de las células infectadas y que genera una variedad de funciones únicas en los b-Coronavirus. En primer lugar, es capaz de bloquear y degradar el mRNA del IFN-b y otros mensajeros endógenos. En segundo lugar, al bloquear la fosforilación de STAT1 e I-kB y la dimerización de IRF3, inhibe las vías de transducción de señales que involucran IRF, STAT y NF-k Ello afecta también a la progresión del ciclo celular sin inducir apoptosis, favoreciendo la supervivencia viral.
La nsp3, codificada en el gran ORF1 y que genera una proteína de 200 kDa con varios dominios funcionales diferentes. Estos incluyen un dominio de unión de poli (ADP-ribosa), de proteasa (papain-like protease, PLP), otro deubiquitinasa y un último de ISGylase. La función PLP de nsp3 es la que antagoniza las respuestas IFN de tipo I, impidiendo la fosforilación, dimerización e importe al núcleo de IRF3. De forma análoga actúa sobre la vía de represión de NF-k Además, la función ISGylase contrarresta la acción del gen 15 estimulado por interferón (ISG15), que es la ISGilación. Veamos en qué consiste este proceso. La modificación postraduccional de las proteínas es una estrategia importante para la regulación del proteoma celular. La ubiquitina y otros modificadores similares a la ubiquitina como ISG15, a través de su conjugación covalente a proteínas median la regulación de los niveles de esas proteínas, las vías de señalización, el tráfico vesicular y muchos otros procesos celulares. ISG15 se codifica y se expresa como una proteína precursora de 17 kDa que se procesa proteolíticamente en su extremo C-ter para exponer una secuencia de aminoácidos, idéntica a la secuencia C-ter de la ubiquitina, y sus adiciones covalentes a las proteínas son bastante similares. Por este mecanismo se regula a la baja la vida media de las proteínas virales, disminuyendo su capacidad de ensamblaje y la viabilidad de los mismos. De forma contraria este proceso afecta aumentando la vida media de muchos genes regulados al alza como respuesta de IRF. Por este motivo esta actividad ISGylase parece una estrategia acertada de propagación viral.
Morales DJ, J Mol Biol 2013
Las otras nsp4 a nsp6 bloquean directamente la actividad antiviral que la célula debería poner en marcha inhibiendo la señalización del IFN. Algunas formas descritas por SARS-CoVs de antagonizar a los IFN de tipo I es mediante la inactivación de la traducción del hospedador; la degradación de sus mARN; interactuar negativamente con STING; antagonizar la señalización de IFN inducida por MAVS/RIG-I. Así mismo, también lo consiguen bloqueando la expresión de citocinas proinflamatorias y la generación de las vesículas que son necesarias para la formación de autofagosomas y el incremento de la presentación de péptidos virales.
Por último, la nsp16 es una 2’O metiltransferasa (2’‐O‐MTasa) que bloquea muy selectivamente la señalización a través de MDA5, un tipo de RIG-like receptor.
Tortura AL, Curr Opinion Virol 2012
La afectación de las células presentadoras de antígeno
Pero aquí non acaba la cosa, tanto los factores virales antes descritos como los del huésped afectan la virulencia de las enfermedades por coronavirus en animales. Un ejemplo de ello es que tanto Covid-19 como SARS se caracterizan por una respuesta inflamatoria exacerbada y, de hecho, la carga viral no está correlacionada con el empeoramiento de síntomas. En este contexto es vital conocer el mecanismo que desencadena que la respuesta inflamatoria del hospedador, con sobreexpresiones de citocinas, se convierta en una causa principal de daño pulmonar y posterior mortalidad en los casos severos de Covid-19. Las dianas principales de la infección por los SARS-CoV son las células epiteliales ciliadas de la vía aérea y los pneumocitos alveolares de tipo II.
Pero también se ha demostrado que los SARS-CoV usan DC-SIGN y L-SIGN como correceptores para la entrada. Es de señalar que cada uno estos receptores son muy expresados en algunos tipos de células presentadoras de antígenos inmaduras (APC), sobre todo células dendríticas (DC) y podría aumentar el tropismo viral y por consiguiente contribuir aún más en la inmunopatogenia de la enfermedad. La alta infectividad viral en APC inmaduras indica que hay muchos PRR en ellas y que el virus puede superar la respuesta antiviral inicial. Pero también nos revela que la maduración de las APC no sólo conlleva una bajada dramática del número de PRR sino la aparición de otros mecanismos de restricción que evitan la replicación de virus. Estas y otras APC actúan como primera defensa contra la infección viral al estimular la vigilancia inmunológica, la defensa antiviral intrínseca y el puente entre la inmunidad innata y adaptativa participando en el reclutamiento de los linfocitos de memoria y de células efectoras al sitio de infección. Además, estas APC profesionales también son una fuente importante de interferones tipo I. Así, el efecto citopático del coronavirus es un mecanismo indirecto de sobrestimulación de APC y se asocia también con la inducción de inflamación y la secreción masiva de citocinas en los pulmones como IL-1, IL-6, IL-8, TNF-a y CXCL-10 que pueden provocar daños en los tejidos y favorecer la diseminación del virus sistémica.
Por otro lado, el mecanismo de escape inmune a través de la afectación de APCs resultaría en un aclaramiento viral ineficiente, lo que puede explicar la alta patogenicidad y las manifestaciones clínicas visto en Covid-19, MERS y SARS. Luego que las APC funcionen adecuadamente es crítico para la mitigar la infección y limitar el desarrollo de la enfermedad. En este sentido, existen tres posibles candidatos terapéuticos sobre la mesa, baricitinib, fedratinib y ruxolitinib que son inhibidores potentes y selectivos de JAK aprobados para indicaciones como artritis reumatoide y mielofibrosis Los tres son potentes antiinflamatorios que inhiben la señalización JAK-STAT de los receptores de citocinas. Desde el punto de vista técnico y de desarrollo de fármacos, esta permisividad de APC sobre coronavirus nos va a ayudar en el screening in vitro de potenciales agentes terapéuticos.
Consideraciones finales
En algunos individuos, la presencia del coronavirus en las vías respiratorias va a ser la puerta de entrada a otros microorganismos oportunistas que podrían ocasionar estas neumonías severas. Esta puerta se abre por el efecto citopático directo del virus sobre las células epiteliales del tracto respiratorio y por el daño tisular local que la respuesta inflamatoria va a ocasionar. Si a esto le añadimos un sistema inmunitario ineficiente debido a los mecanismos de inmunoevasión del coronavirus, tenemos todas las papeletas para convertir a Covid-19 en una enfermedad especialmente contagiosa, perdurable en el individuo y en algunos casos grave. Los mecanismos de escape viral e inmunopatogenia no son únicos de los coronavirus y han sido descritas muchas formas diferentes de interferir en la señalización de citocinas y de sus mecanismos de transducción. Además, otros virus de las familias de adenovirus, herpes, papiloma y poxvirus, entre otros interfieren en la actividad del proteasoma y el proceso celular dedicado a la presentación antigénica o de aparición del MHC en la membrana. Otros codifican para citocinas inmunosupresoras o cambios de fenotipos de los linfocitos helper (como el shift Th1 a Th2, poco eficiente para parásitos intracelulares). Generalmente son grandes virus de DNA y aquí los coronavirus representan una buena excepción a esta regla.
Hasta el momento, Covid-19 se está comportando como un enemigo de la especie humana, con unas tasas de letalidad difícilmente asumibles y que está poniendo en jaque el movimiento de personas, la globalización y la economía mundial. Sin ninguna duda, sus efectos sobre países en vías de desarrollo serán todavía peores. Por ello, como enemigo que es de todos nosotros, requiere un esfuerzo global para combatirlo. Destinemos los recursos necesarios para comprender mejor la patogenia, desarrollar antivirales y medidas de soporte más efectivas y luchemos por conseguir la vacuna preventiva. Tomemos las medidas oportunas para contener la diseminación de la enfermedad y protejamos a los más sensibles o susceptibles a sucumbir ante el virus, pero con cautela y sin alarmar innecesariamente a la población. Pero eso sí, no deberíamos permitir que Covid-19 se convierta en la pandemia del siglo XXI como sucedió con la gripe de 1918.
Referencias
Cong, Y., Hart, B. J., Gross, R., Zhou, H., Frieman, M., Bollinger, L., … Holbrook, M. R. (2018). MERS-CoV pathogenesis and antiviral efficacy of licensed drugs in human monocyte-derived antigen-presenting cells. PLoS ONE, 13(3), 1–17. https://doi.org/10.1371/journal.pone.0194868
Morales, D. J., & Lenschow, D. J. (2013). The antiviral activities of ISG15. Journal of Molecular Biology, 425(24), 4995–5008. https://doi.org/10.1016/j.jmb.2013.09.041
Cui, J., Li, F., & Shi, Z. L. (2019). Origin and evolution of pathogenic coronaviruses. Nature Reviews Microbiology, 17(3), 181–192. https://doi.org/10.1038/s41579-018-0118-9
Chen, Y., Liu, Q., & Guo, D. (2020). Emerging coronaviruses: Genome structure, replication, and pathogenesis. Journal of Medical Virology, (January), 418–423. https://doi.org/10.1002/jmv.25681
Stebbing, J., Phelan, A., Griffin, I., Tucker, C., Oechsle, O., Smith, D., & Richardson, P. (2020). COVID-19: combining antiviral and anti-inflammatory treatments. The Lancet Infectious Diseases, 2019(20), 2019–2020. https://doi.org/10.1016/S1473-3099(20)30132-8
Poulter, N. (2020). Lower Blood Pressure in South Asia? Trial Evidence. New England Journal of Medicine, 382(8), 758–760. https://doi.org/10.1056/NEJMe1917479
Zhu, N., Zhang, D., Wang, W., Li, X., Yang, B., Song, J., … Tan, W. (2020). A Novel Coronavirus from Patients with Pneumonia in China, 2019. New England Journal of Medicine, 727–733. https://doi.org/10.1056/nejmoa2001017
Totura, A. L., & Baric, R. S. (2012). SARS coronavirus pathogenesis: Host innate immune responses and viral antagonism of interferon. Current Opinion in Virology, 2(3), 264–275. https://doi.org/10.1016/j.coviro.2012.04.004
Li, F. (2013). Receptor recognition and cross-species infections of SARS coronavirus. Antiviral Research, 100(1), 246–254. https://doi.org/10.1016/j.antiviral.2013.08.014
Frieman, M., & Baric, R. (2008). Mechanisms of Severe Acute Respiratory Syndrome Pathogenesis and Innate Immunomodulation. Microbiology and Molecular Biology Reviews, 72(4), 672–685. https://doi.org/10.1128/mmbr.00015-08
Etiquetas: antiviral, citocina, coronavirus, Inmunopatogenia, interferon
← ¿Por qué un blog sobre inmunología?La tormenta de citocinas y su implicación en la patogenia del virus de la gripe A y del Covid-19 →
Funciona con WordPress / Tema Academica
Por Rafael Sirera en Inmunopatogenia sobre 09/03/2020