ENRIQUE RUBIO GARCIA
Jefe del Servicio de Neurocirugía Valle de Hebron
Profesor Titular de Neurocirugía
Academico de España, Portugal, European Society of Neurosurgery, Word Federation of Neurosurgery.
Investigador del I Carlos III
Veintidós tesis doctorales dirigidas
250 trabajos publicados
Presidente de la academia de Neurocirugía de Barcelona
Academico de Cadiz y Jerez de la Frontera
Acadenico de Honor de Andalucia y Cataluña
log enriquerubio.net
Cuando en medio de la pandemia por coronavirus, disminuye marcadamente el número de infecciones. Es natural que la curiosidad nos lleve a pensar “qué está pasando “.
Las medidas de prevención que estamos utilizando, son las causantes de esta desaparición o mengua las infecciones, como la gripe y otras varias.
y sí lo son, por qué no responden de igual manera a las rigurosas prevenciones que estamos utilizando para los coronavirus.
Hay algo más que se nos escapa y por ello copio este articulo sobre los pocos conocidos virofagos.
Según el Comité Internacional de Taxonomía Vírica, un virus es un biosistema elemental que posee algunas de las propiedades de sistemas vivientes: genoma y capacidad de adaptación a cambios medioambientales. Sin embargo, un virus no puede capturar y almacenar energía libre y no es funcionalmente activo fuera de su célula hospedadora diana. Dicho de otro modo, un virus no tiene metabolismo y, por ello, no es un ser vivo, sino que, como parásito intracelular obligado, toma prestadas las características de los verdaderos seres vivos: desde las bacterias –incluso las que viven en condiciones extremas de temperatura, presión o salinidad- hasta nosotros mismos .
Virófagos. Virus come virus
Por lo tanto, parece lógico pensar que sólo las entidades biológicas con capacidad replicativa y metabolismo propio pueden ser depredadas por virus infecciosos, ¿o no? Pues a juzgar por estudios llevados a cabo en los últimos años en diferentes laboratorios, agentes víricos infecciosos complejos, grandes, muy grandes, recientemente analizados, podrían ser sorprendentes dianas de otros virus “comedores” de virus: los Virófagos. A continuación se describe el artículo recientemente publicado por el autor presente Post en El Cultural:
Algunas de las especies de virus con ADN como genoma más grandes conocidos, como el de la viruela -prácticamente desaparecido a la espera de que la OMS decida qué hacer con las últimas muestras congeladas-, o el de la peste porcina africana, pueden acercarse a los 400 nm de tamaño y prescindir de muchos de los componentes y factores celulares implicados en replicación. Incluso pueden replicar en el citoplasma, lejos del núcleo donde necesitan acudir la mayoría de virus ADN. Sin embargo, hace dos décadas se descubrieron otras entidades víricas gigantes –Giant Viruses o Giruses– capaces de infectar amebas y otros organismos simples. Estas entidades, como las del género Mimivirus, poseen los genomas y cápsidas más grandes y complejos conocidos: hasta 1.2 Mb –más que muchas bacterias- y cerca de la micra –es decir, dentro del rango del microscopio óptico convencional-, respectivamente. Otros ejemplos de virus muy grandes a destacar serían el agente que infecta zooplancton, Virus Cafeteria roenbergensis (CRV), o ciertos destructores de algas de la familia Phycodnaviridae. Aunque no son entidades biológicas independientes, están “a un hervor” de la vida. En su conjunto, todos estos virus grandes se engloban dentro de los denominados Virus ADN Grandes Nucleocitoplásmicos (NCLDV, en inglés). Desde la Universidad del Mediterráneo, en Marsella, el equipo de científicos que participó en la caracterización de los primeros mimivirus anunció en el último congreso europeo –EuroVirology– el proyecto Tara-Girus, un barco-laboratorio que pretende recoger y analizar virus gigantes a lo largo del mundo. Estos giruses, como se verá a continuación, podrían poseer una característica compartida: sus propios parásitos “intravíricos” o virófagos mencionados anteriormente. Por ello, una pregunta que muchos científicos se plantean sería: ¿siguen siendo los NCLDV o giruses organismos no-vivos?
Los virófagos son parásitos conocidos solamente desde hace un par de años. En 2008, el mismo grupo de investigadores que había caracterizado los mimivirus, descubrió el primero de estos virus comedores de virus, apodado Sputnik, en una torre de refrigeración de agua en París. Desde entonces, tal y como auguró Christelle Desnues, microbióloga del Centro Nacional de Investigación Científica de Marsella, nuevos virófagos han ido apareciendo, como el marino Mavirus, capaz de infectar al CRV, predador de especies integrantes del zooplancton ya mencionado. De hecho, en este último caso y según publican en Science científicos de la Universidad de Columbia Británica de Vancouver, Canadá, el genoma del mavirus sería similar al de algunos transposones eucarióticos. Los transposones son elementos genéticos que pueden “saltar” y moverse entre diferentes partes del ADN. Si su inserción se produce en un sitio sensible, pueden producir mutaciones incompatibles con la viabilidad celular o, como en el caso que nos ocupa, vírica. Quizá, ya puestos a especular, algunos transposones pudieran haber derivado de antiguos virófagos pasando a ser elementos utilizados por células superiores como protección frente a determinados giruses. Extrapolando estos resultados, además de permitir proteger a las especies de zooplancton, virus anti-virus podrían, en un futuro, ser utilizados en terapias en seres superiores -¿por qué no en humanos, tal y como se pensó con los bacteriófagos, virus infecciosos en bacterias, antes del desarrollo de los antibióticos?
El último de los virófagos descubierto acaba de ser descrito en las prestigiosas Proceedings of the National Academy of Sciences y Nature. El nuevo miembro de este grupo de parásitos se encontró por casualidad en el inhóspito Lago Orgánico del Éste Antártico mientras se estudiaban otras formas biológicas. De hecho, los autores del trabajo -Escuela de Ciencias Biotecnológicas y Biomoleculares de la Universidad de Nueva Gales del Sur, en Australia-, coordinados por Ricardo Cavicchioli, sugieren que estas formas orgánicas son más frecuentes de lo que se pensaba. El virófago se denomina OLV (Virófago del Lago Orgánico).
Tal y como se comentó anteriormente, estos virus depredan virus gigantes como sería el caso de los ficodnavirus. Al atacar a estos giruses, OLV estaría protegiendo a sus víctimas, esto es, a diversas especies de algas. De hecho, el genoma de OLV se identificó dentro de la secuencia de su hospedador. Para Cavicchioli, por lo tanto, OLV podría estar colaborando con la supervivencia de algunas especies de algas antárticas –al menos durante los meses del deshielo-.
El hecho de que todos los virus que actúan como hospedadores de virófagos pertenezcan al grupo de los NCLDV, parece significativo de la complejidad estructural y funcional de estas últimas entidades. Finalmente señalar que, además de los extraordinarios lagos antárticos, genes que codifican proteínas virales –principalmente de la cápsida- han sido encontrados dentro de hospedadores de los más diversos ambientes, como un lago salino de las islas Galápagos, un estuario de Nueva Jersey, en EE.UU., u otro lago en Panamá, apoyando nuevamente la idea, según comenta Virginia Gewin en Nature, que la expansión de estos comedores de virus podría ser amplia.
Tal y como se ha mostrado, nuevas entidades infecciosas capaces de atacar los organismos más variados están emergiendo incluso de los entornos más inhóspitos, como, por ejemplo, los lagos antárticos. Y mientras todo esto ocurre, el origen y evolución de los primeros virus sigue siendo un misterio. Tres parecen ser las opciones más plausibles, no excluyentes entre sí: evolución retrógrada, por la cual, algún virus complejo podría haber sido originariamente una célula pequeña, simple –procariota probablemente- que tras parasitar otra de mayor complejidad fue especializándose y perdiendo autonomía. Otra opción apunta a un posible material celular que habría escapado al control de la división para perpetuarse de forma independiente. Por último, la tercera hipótesis –que no tiene por qué ser la menos probable- hablaría de una evolución independiente de los virus desde un mundo primitivo rico en ARN dentro de un océano prebiótico. Algunas de estas moléculas de ARN aprenderían a perpetuarse en el interior de las primeras células a medida que iban surgiendo.
¿Dónde encajarían los nuevos virófagos en toda esta vorágine infectivo-biológica? Pues no se sabe. Quizás sean el resultado lógico de una evolución reduccionista a varias bandas: células simples que pasarían a virus gigantes y parásitos suyos previos que co-evolucionarían a virófagos. Sea como fuere, el descubrimiento y posterior caracterización de nuevas formas infecciosas abre nuevas perspectivas en materia de evolución –tal y como ya ocurrió con los retrovirus, ribozimas y priones– y desdibujan aún más la delicada frontera entre la vida y lo simplemente orgánico.
Para terminar, podríamos explicar la disminución de enfermedades infecciosas en tiempo de pandemia por coronavirus, Porque estos bichos se comen entre sí . lo mas probable que la cosa no sea así, ´pero y si lo es.
El Aplidin, el antiviral de PharmaMar, es cien veces más potente que el tratamiento que se está usando para el covid-19
Si el Aplidin, es un antivirus potente como afirma su creador, estamos salvados .
Tiene fuerte efecto contra los virus .
Esto es producto tiene de la gran preocupación que causa esta epidemia en el mundo y de que miles de investigadores están buscando una solución , porque nos va en ello la vida .
Siempre tenemos el miedo de la fake new. pero tampoco se puede dudar de todo .
Este medicamento era ya conocido en el tratamiento de los tumores. La plitidepsina (aplidina): no ataca a una proteína del virus, sino que bloquear la molécula diana que está dentro de nuestras células (eEF1A), que no muta, que no cambia, y que es necesaria para el SARS-CoV-2, sea cual sea la cepa, actue.
Es ilusionante y por eso copio este articulo
Un estudio internacional, publicado en la prestigiosa revista «Science», confirma el alto índice terapéutico de la plitidepsina, un fármaco contra el cáncer que bloquea el virus independientemente de cuál sea su cepa
El fármaco contra el covid-19 de la biotecnológica gallega PharmaMar (el Aplidin) acaba de recibir un importante espaldarazo: un equipo de investigadores internacionales, liderados por el virólogo español Adolfo García-Sastre, del Hospital Monte Sinaí de Nueva York, ha publicado en la prestigiosa revista Science los primeros resultados de un estudio sobre la plitidepsina que demuestran que este fármaco, concebido inicialmente para tratar el cáncer, es cien veces más potente que el remdesivir, actualmente el único antiviral aprobado para tratar la enfermedad que provoca el SARS-CoV-2.
«Este trabajo confirma tanto la potente actividad como el alto índice terapéutico de la plitidepsina que, por su especial mecanismo de acción, inhibe este coronavirus independientemente de cual sea su mutación en su proteína “S” (cepas británica, sudafricana, brasileña o las nuevas variantes que han salido recientemente en Dinamarca)», celebra el presidente del grupo, José María Fernández Sousa. La farmacéutica trabaja ya con agencias del medicamento para empezar el ensayo de fase 3, que se va a desarrollar en varios países. Teniendo en cuenta los tiempos que requieren estos estudios y lo que tardan los reguladores en dar su aprobación, el fármaco podría estar listo a mediados de este año.
«El virus, para replicarse, para hacerse copias, necesita algo similar a una máquina fotocopiadora, que sería esta proteína eEF1A -explican la biotecnológica-. Nosotros lo que hacemos es apagarla. Por tanto, el virus no puede hacer copias y la infección se paraliza». Eso sí, para que sea útil, el medicamento debe ser administrado en el momento adecuado, en la fase viral de la enfermedad, cuando el agente infeccioso aún está en el cuerpo haciendo daño. No se puede tardar mucho. «Es como cuando hay un incendio en una casa. Para salvarla, los bomberos tienen que llegar cuando el fuego está empezando. Las llamas tienen que ser atajadas al principio».
DISMINUCIÓN DE LAS INFECCIONES DURANTE LA EPIDEMIA DEL CORONAVIRUS
Absolutamente inesperado es la bajada del número de casos de otras enfermedades de carácter infeccioso durante la pandemia del SARS-CoV-2 Podriamos decir que este efecto colateral nos alegra enormemente, pero al mismo tiempo nos sorprende.
Adelanto que la explicación de este hecho, no es simple, que nadie se entusiasme, pero este acontecimiento es espectacular .
Gracias a los datos que arroja el Informe semanal de Vigilancia del Instituto de Salud Carlos III, podemos saber la evolución de decenas de enfermedades infecciosas a lo largo de 2020 con respecto a años anteriores en las mismas fechas. Desde hace décadas, los médicos en España están obligados a notificar a la Red Nacional de Vigilancia Epidemiológica diversas enfermedades infecciosas cuando las detectan durante la práctica clínica. Cada semana, se publica el citado boletín dirigido principalmente a profesionales sanitarios e investigadores, aunque también al público en general, con los resultados de esta vigilancia de enfermedades transmisibles de declaración obligatoria (EDO).
Esta lista está compuesta por un total 60 enfermedades infecciosas (gripe, tuberculosis, tétanos, varicela, sífilis…) y se clasifican en enfermedades de transmisión alimentaria, parenteral, respiratoria, vectorial, zoonótica (procedente de animales), sexual y en aquellas que se podrían evitar con vacunaciones. Algunas de ellas (alrededor de una veintena) se registran semanalmente y otras son de declaración urgente (fiebre amarilla, peste, fiebres hemorrágicas…). La idea principal tras este boletín semanal es alertar rápidamente cuando se detecta un aumento inesperado de casos de ciertas enfermedades infecciosas o cuando aparece un brote por cualquier causa.
Los datos son rotundos: prácticamente todas las enfermedades infecciosas documentadas (salvo la COVID-19) han disminuido de forma contundente su incidencia en 2020. Desde las más frecuentes como la gripe, hasta aquellas con una frecuencia muy reducida como el tétanos, la brucelosis o la rubéola. Aunque, como avisan en el boletín, es posible que por la pandemia de SARS-CoV-2 «los cero casos se deban a falta de notificación en algunas comunidades autónomas», la tendencia general es clara y uniforme.
Un análisis de los datos epidemiológicos de las EDO en los últimos cinco años (2016-2020), acumulados en la semana 35, refleja dos tendencias generales diferentes. Por un lado, se encuentran los casos de múltiples enfermedades infecciosas que ya iban en descenso desde 2016 y que han disminuido bruscamente en 2020. En este grupo encontramos a la tos ferina, la infección gonocócica o gonorrea, la tuberculosis, la varicela, la hepatitis A, la legionelosis, la hepatitis B, la brucelosis o el tétanos. También existe un descenso de los casos de gripe, aunque ligero, ya que la pandemia de coronavirus comenzó en España cuando la epidemia de gripe daba sus últimos coletazos.
Por otro lado, se registran casos de diversas enfermedades infecciosas que se mantenían en el tiempo o incluso iban en aumento y que han disminuido notablemente en 2020. En este grupo encontramos a la sífilis, el paludismo, la shigelosis, el sarampión, la fiebre tifoidea y paratifoidea, la parotiditis y la enfermedad meningocócica.
Y esto no ocurre solamente en España sino que en el mundo que pública ocurre también
Así lo pone de manifiesto un trabajo publicado recientemente en la revista «The Pediatric Infectious Disease Journal» realizado en el Royal Children’s Hospital de Melbourne (Australia), y según el cual entre el 1 de abril y el 31 de mayo de 2020 (periodo en el que las medidas anticoronavirus ya estaban presentes), se produjo una disminución en los ingresos hospitalarios relacionados con infecciones comparado con el mismo lapso de 2019. Según estos datos, en ese tiempo hubo una reducción drástica de 1.264 pacientes en comparación con el mismo período de 2019 a 2005, así como una bajada proporcional en las admisiones relacionadas con las infecciones del 41% al 30% (818 casos vs 375).
El descenso más significativo, del 91%, tuvo que ver con el crup o laringotraqueobronquitis (una infección de las vías respiratorias superiores que causa dificultad respiratoria y tos en niños) que pasó de 22 casos a tan sólo dos, seguido del 86% de la bronquiolitis que bajó a 30 casos frente a 212.
Además confirma que, aunque aún no existen datos estadísticos al respecto, en nuestro país «ha habido menos ingresos de neumonías y enfermedades por otros virus y bacterias respiratorias durante el periodo Covid», si bien apunta que esto podría también deberse al «cambio de actitud de la población de no acudir a las urgencias por cuadros más leves».
Así lo cree también María García Onieva, pediatra de atención primaria y secretaria del Comité Ejecutivo de la Asociación Española de Pediatría (AEP), quien subraya que «se han publicado ya resultados de la epidemia de gripe en países de esas latitudes y se ha visto que han disminuido muy notablemente los casos de gripe confirmados por laboratorio: un 93% en Australia, en Chile se han reportado hasta 18 veces menos casos que el año anterior y en otros países como Argentina o Nueva Zelanda el descenso también ha sido importantísimo». «Aunque a priori no podemos dar una cifra de la bajada de infecciones que esperamos en España por gripe este año, por efecto del confinamiento y las medidas anticovid está claro que vamos a ver una reducción en todas las enfermedades respiratorias», asegura Menéndez. A lo que además, añade, hay que sumarle el más que probable aumento en las tasas de vacunación de este año, lo que contribuirá más aún a frenar el número de contagios.
Y no sólo en los casos de gripe, «los de bronquiolitis han sufrido también un descenso, está menos presente que otros años. Existen evidencias de que han disminuido el número de enfermedades infecciosas, sobre todo las respiratorias, pero también las gastrointestinales como consecuencia de las medidas tomadas por la pandemia –añade Onieva–.
Otras que ha mejorado mucho son las enfermedades que se transmiten dentro de los hospitales. «Ahora todos los sanitarios vamos con mascarilla y hacemos todas las medidas de higiene, lo que se ha materializado en un descenso, aún no cuantificado oficialmente, de las infecciones nosocomiales, pero sí que lo hemos notado –asegura Almirante–. A día de hoy se han reducido alrededor de un 50% estas infecciones. Esto es así». «En los hospitales el uso de la mascarilla ha llegado para quedarse –corrobora Menéndez–. Antes no la utilizábamo siempre, sólo en el pico gripal (no así el lavado de manos que sí que se hacía), pero ahora se lleva todo el tiempo y con todo tipo de pacientes».
También las infecciones de transmisión sexual (ITS) se vieron reducidas en un primer momento como consecuencia del coronavirus, pero en este caso por el confinamiento obligatorio. Nos referimos a sífilis, gonococia clamidia o el herpes genital, terminando así con años de cifras al alza, un repunte en casi todas las infecciones de este tipo sostenido desde 2003. Valga como ejemplo lo que cuenta el doctor Almirante: «Nosotros tenemos clínica específica de ITS y la pudimos cerrar durante los tres meses del confinamiento. Se produjo un descenso de la actividad de más del 90%, lo que es lógico. Ahora hay alrededor del 50% de la actividad».
«La idea es que hay menos de todo, menos infecciones del tipo mononucleosis, enfermedad boca-mano-pie o impétigo contagioso (causada por el estafilococo aureus) y menos infectaciones (de piojos o escabiosis) porque necesitan de un contacto directo y mantenido y obviamente esto ya no es así o será más difícil que se de. Habrá un descenso de la probabilidad de contagio del resto de infecciones», concluye Gloria Garnacho, miembro del Grupo de Tricología de la Academia Española de Dermatología y Venereología (AEDV).
Las posibles razones para este panorama favorable de las enfermedades infecciosas, al margen de la COVID-19, son numerosas y muy variadas. Por ejemplo, las diversas acciones dirigidas a reducir los contagios por coronavirus podrían haber sido útiles para disminuir también el contagio de enfermedades infecciosas respiratorias como la tuberculosis (cuyos casos se han reducido al 60% con respecto a 2019). Por otra parte, el confinamiento limitó las interacciones entre no convivientes y es probable que esto haya influido en una menor incidencia de las enfermedades de transmisión sexual y de enfermedades que se pueden prevenir por vacunación (como por ejemplo el sarampión). También es posible que algunos casos de estas enfermedades infecciosas no se hayan detectado por el miedo a ir a los centros de salud y a los hospitales durante los peores momentos de la epidemia en marzo-abril.
A pesar de la clara dinámica general de las enfermedades infecciosas en 2020, existen llamativas excepciones: las enfermedades infecciosas de origen tropical como la fiebre hemorrágica de Crimea-Congo y la fiebre del Nilo Occidental han ido a más en 2020. Los primeros casos conocidos de fiebre hemorrágica en España, transmitida por garrapatas, se remontan a 2016. Desde entonces hasta ahora, se han confirmado 6 casos de esta fiebre hemorrágica (3 muertes) en nuestro país, dos de los cuales han ocurrido en 2020. El porcentaje de las garrapatas en España que poseen el virus ha aumentado notablemente en los últimos años.
La situación de la fiebre del Nilo (transmitida por mosquitos) es más preocupante, pues este año ha ocurrido el mayor brote registrado en nuestro país, con decenas de casos confirmados y siete fallecidos. No se detectó ningún caso de esta enfermedad en humanos tanto en 2017 como en 2019 y solo un caso en 2018. El aumento drástico de los casos de la fiebre del Nilo podría deberse a la proliferación de mosquitos en diferentes partes de la geografía española por el confinamiento (con una actuación menor para el control de estos insectos) y una primavera favorable para ello.
Este hecho espectacular de la desaparición un menguado de las infecciones en tiempo del coronavirus , necesitan para explicar la algo más que la prevención qué estamos utilizando. Esto es más difícil aquí intervienen más factores de los que estamos utilizando.
«The Pediatric Infectious Disease Journal» realizado en el Royal Children’s Hospital de Melbourne (Australia) realizado en el Royal Children’s Hospital de Melbourne (Australia)
LA PRIMERA IMAGEN REAL DEL CORONAVIRUS EN 3 DIMENSIONES
Esta claro que los avances en tecnología, nos permiten ver cosas y pequeñeces hasta ahora inimaginables
La imagen real del coronavirus en tres dimensiones me parece un regalo.
Ni siquiera imagino, como se puede hacer estas cosas. No se pueden ver en un microscopio tampoco en un modelo computerizado. Lo que un equipo de investigadores en tres países ha logrado es la primera imagen real y en tres dimensiones del SARS-CoV-2, un avance que puede ayudar a los científicos a luchar contra él.
«Es lo más cercano a mostrar la apariencia real del virus que hemos logrado hasta ahora. Con la tecnología actual, no se puede mostrar una imagen más real”
Para obtener la imagen se usó la técnica de tomografía crioelectrónica, en el que la muestra congelada se va escaneando desde distintos ángulos usando un microscopio electrónico
Primera imagen real y en tres dimensiones del coronavirus. NANOGRAPHICS EFE
No es una foto (no se puede fotografiar a un virus), pero tampoco en un modelo computerizado. Lo que un equipo de investigadores en tres países ha logrado es la primera imagen real y en tres dimensiones del SARS-CoV-2, un avance que puede ayudar a los científicos a luchar contra él.
«Es lo más cercano a mostrar la apariencia real del virus que hemos logrado hasta ahora. Con la tecnología actual, no se puede mostrar una imagen más real», resume para Efe Peter Mindek, director de tecnología de Nanographics, la empresa austríaca que ha creado la imagen, junto a centros universitarios de China y Arabia Saudí.
Para obtener la imagen, un objeto esférico del que surgen las famosas espículas, se usó la técnica de tomografía crioelectrónica, en el que la muestra congelada se va escaneando desde distintos ángulos usando un microscopio electrónico.
Los datos obtenidos se transforman en imágenes tridimensionales usando algoritmos.
Primera imagen real y en tres dimensiones del coronavirus.NANOGRAPHICSEFE
La tomografía se realizó en la Universidad Tsinghua, en China, y los datos obtenidos fueron segmentados luego por expertos de la Universidad de Ciencia y Tecnología Rey Abdalá.
Finalmente, Nanographics, fundada por científicos de la Universidad Técnica de Viena, eliminó el ruido de la imagen original, la renderizó y le asignó propiedades ópticas y colores.
Mindek recuerda que un virus es más pequeño que la longitud de onda de la luz visible, por lo que, por ejemplo, ni siquiera tiene color.
COLOR FALSO PERO FORMA REAL
Por eso, los tonos rosas y azules usados en la imagen son, como él dice, «falsos», con el propósito de ayudar a representar mejor la forma y las distintas partes del virus.
Lo que sí es real es la forma del virus, algo que tiene mucha importancia para los científicos que buscan formas de combatirlo. «Los científicos que investigan vacunas y curas necesitan saber la forma de las moléculas. Si lo ven en 3D, es más fácil saber cómo funcionan», explica Mindek.
Aunque hemos oído hablar muchas veces de las gotitas que transportan los Virus SARS-CoV-2 en esta terrible enfermedad, está claro el tamaño de la gota el peso de la misma la duración en la atmósfera que nos rodea y el tiempo de exposición son importantes.
No solo importa el tamaño de las gotas: los flujos y partículas aéreas están sometidos a muchos fenómenos. Solo así se explica el ‘éxito’ del coronavirus.
Ya tenemos algunas cosas claras.
Las personas agrupadas en un lugar con poca ventilación son las fuentes mayor de contagio
Los hospitales, la residencia de ancianos y los grupos de persona aislado en un lugar con poca ventilación se contagian sistemáticamente y supone un porcentaje más de 50% de los contagios.Los sanitarios y mayores lo tienen mal.
Sentados en la calle con Con 2 metros de distancia entre personas y mascarilla y sobre todo si hay una buena ventilación. el aire de la calle, la infección es claramente menor,
Viene el problema, de si los virus se transmiten además de otras formas que no conocemos. Lo que sí vemos con claridad es que tras una festividad donde la gente se agrupa mucho el contagio está asegurado.
SARS-CoV-2Mantener la distancia es importante, pero hay que tener en cuenta otros factores de protección para salvarse del contagio.
Un equipo internacional y multidisciplinar coordinado por Julian W. Tang, de la Universidad de Leicester, y Stephanie J. Dancer, de la Universidad de Edimburgo, explican y refutan en el número de enero de The Journal of Hospital Infectionseis mitos o malentendidos sobre la difusión del coronavirus. Entre los firmantes figuran expertos en aerosoles, ventilación, ingeniería, física, epidemiología, virología y medicina clínica de varias universidades de todo el mundo.
ESTOS ANCIANOS ESTAN MAS ESPUESTOS A CONTAGIOAQUI ES MAS DIFICIL
Comprender los mecanismos de transmisión del SARS-CoV-2 es clave para prevenir su propagación, pero hasta mediados del año pasado no quedó claro el poder aerotransportado del virus y aún hoy sigue habiendo confusión sobre la infectividad de aerosoles, gotas, gotículas y partículas.
No hay duda -dicen- de que el SARS-CoV-2 se transmite a través de una gama variable de tamaños de partículas en el aire sujetas a parámetros de ventilación y de comportamiento humano. Reconocen sin embargo la escasez de pruebas sólidas sobre la infectividad en algunos escenarios, como el de los polémicos fómites o las partículas muy pequeñas, y la dificultad, por sus fallos y su coste, de seguir los contagios a través de adecuadas secuenciaciones genómicas que detecten el origen exacto en una superficie, una mano o un aerosol.
Cada uno de los mitos que analizan se deriva de estudios con cierta evidencia y de su experiencia en cada una de las disciplinas científicas. “Esperamos facilitar la comprensión de por qué algunas declaraciones comunes están obsoletas y por qué las pruebas actuales apuntan en una dirección diferente”.
Mito 1: «Los aerosoles son gotas con un diámetro de 5 micrómetros (µm) o menos»
Este mito se originó a partir de una definición históricamente incorrecta y recogida por la Organización Mundial de la Salud: «… las gotas de <5 µm de diámetro se conocen como núcleos de gotas o aerosoles».
Las gotas respiratorias, formadas a partir de secreciones respiratorias y saliva, se emiten a través del habla, las toses, los estornudos e incluso la respiración. Sus diámetros abarcan un espectro de <1 a >100 µm. Las más pequeños se secan rápidamente al 20-40% de su diámetro original, dejando residuos llamados «núcleos de gotas», que la mayoría de los médicos creen que son sinónimos de «aerosoles».
Con una amplia gama de diámetros, esas gotas exhaladas pueden permanecer suspendidas en el aire y considerarse aerosoles. No es posible especificar un punto de corte para el diámetro de las partículas en el aire porque la capacidad de una partícula para permanecer suspendida depende de muchos factores distintos del tamaño, incluyendo la fuerza con la que se expulsan, y las características del flujo de aire circundante (velocidad, turbulencia, dirección, temperatura y humedad relativa).
Dependiendo de esas condiciones, muchas partículas que previamente habrían sido clasificadas como ‘grandes’ (diámetro >5 µm) pueden viajar mucho más lejos que la distancia ‘mítica’ de 1-2 metros, dentro de la cual se afirma que tales partículas caen al suelo. Así que teniendo esto en cuenta, incluso las partículas grandes también pueden comportarse como ‘aerosoles’ tradicionales. Tanto los ‘aerosoles’ como las ‘gotas’ deben considerarse como extremos de un rango de tamaño para el que su patrón aerotransportadovariará en función de las condiciones ambientales.
Los autores definen las ‘gotas’ como partículas que caen al suelo (o cualquier superficie incluyendo las verticales) bajo la influencia de la gravedad o el impulso del aire exhalado de una persona infectada, y los ‘aerosoles’ como partículas que permanecen suspendidas debido al tamaño o las condiciones ambientales. El término ‘partículas’ designa tanto gotas como aerosoles.
A efectos de la transmisión, un umbral de tamaño más racional para distinguir las gotas de los aerosoles, en términos de su comportamiento físico y de su vía de exposición, es de 100 µm.
Mito 2: «Todas las partículas de más de 5 µm caen a 1-2 metros de la fuente»
Es una afirmación muy repetida, pero científicamente falsa. Las partículas exhaladas de 5-10 µm de diámetro caen lentamente al suelo bajo la influencia de la gravedad en el aire interior. Esto supone de 8 a 30 minutos desde una altura de 1,5 m. Sin embargo, la mayoría de las habitaciones tienen corrientes de aire ambiente de 0,1-0,2 m/s, lo que significa que estas partículas son demasiado pequeñas para asentarse en el suelo dentro de esa distancia de 1-2 m de la fuente.
Una gota debe ser mayor que 50-100 µm para tener una alta probabilidad de aterrizaje a esos 1-2 m de la fuente emisora en interiores. Los flujos de aire pueden extender este tiempo de suspensión por más tiempo. Y esas gotas de más de 50-100 µm pueden recorrer más de dos metros impulsadas por toses o estornudos.
Las partículas que son demasiado pequeñas para asentarse rápidamente por la gravedad pueden moverse hacia arriba en el penacho térmico de una persona, la columna de aire caliente en movimiento ascendente producida por el calor corporal. Estas partículas pueden verse influenciadas por otros flujos de aire generados por la ventilación, el tráfico de personas, los movimientos de las puertas y los flujos convectivos (por ejemplo, corrientes de aire producidas por equipos eléctricos y cuerpos calientes), antes de ser finalmente inhaladas. Tales flujos son especialmente importantes para las partículas de <5-10 µm, que se pueden transportar a distancias de más de dos metros.
En el aire quieto, las partículas de diferentes tamaños tienen diferentes tiempos de sedimentación en función de leyes físicas, como la de Stokes. Así, los cálculos muestran que incluso las partículas con un diámetro de alrededor de 50 µm tardarán unos 20 segundos en asentarse a partir de una altura de 1,5 m y deben considerarse como aerosoles. Las ligeras turbulencias en las salas de hospitales pueden hacer que partículas de ese tamaño aguanten más tiempo en el aire y sean capaces de viajar más de 2 metros desde la fuente.
El período de tiempo clínicamente relevante para las partículas suspendidas en el aire depende de la ventilación. Los sistemas de ventilación de un hospital suministran aire limpio de forma continua. Si la habitación tiene una tasa de intercambio de aire de 6 cambios por hora (ACH), entonces la duración de interés es de 10-30 minutos. Si la tasa es de 12 ACH, entonces la duración baja a 5-15 minutos. Si el hospital no tiene sistemas de ventilación mecánica y en ausencia de ventanas o puertas abiertas, las partículas podrían tardar horas en asentarse en el suelo.
Mito 3: «Si es de corto alcance, entonces no puede ser aerosol»
La distancia social de 1-2 m, distancia conversacional, es la que define el corto y el largo alcance. Comúnmente se piensa que la transmisión de largo alcance es una prueba de transmisión en el aire, pero la ausencia de transmisión de largo alcance detectable no excluye la transmisión aérea.
El contagio del agente infeccioso por medio de la inhalación puede ocurrir a cualquier distancia, si bien es más probable que ocurra a corta distancia pues los aerosoles se concentran cerca de la fuente. Basta observar cómo se disipa el humo de un fumador. Un fenómeno similar se puede experimentar por el olfato, por ejemplo, si se está lo suficientemente cerca de alguien que ha comido ajo o bebido alcohol o se ha perfumado: el olor se desvanece a medida que uno se aleja. Sin embargo, si la respiración exhalada sigue difuminando olores perceptibles, entonces también se puede estar inhalando cualquier virus presente en ese aliento exhalado. Muchos brotes son difíciles de explicar sin esa inhalación de SARS-CoV-2 aerosolizado.
Los aerosoles se concentran obviamente a corta distancia del emisor infeccioso (<1 m): desde gotas grandes balísticas hasta aerosoles diminutos. Que haya transmisión en rangos mayores (más allá de 1-2 m) depende de varios parámetros: cantidad de viriones en el aire producidos por la fuente; distribución de viriones transportados por diferentes tamaños de partículas; patrones de flujo de aire en el entorno; tasa de descomposición de la infectividad del virus; dosis infecciosa necesaria para causar una infección; dilución del inóculo a distancia; y eliminación oportuna por aire fresco, ventilación o limpieza del aire.
El riesgo de transmisión de mayor alcance (>2 m) es menor en comparación con el de corta distancia (<1 m), pero puede ocurrir. Desafortunadamente, los eventos de transmisión de gran alcance pueden ser muy difíciles de probar cuando ese patógeno ya está muy extendido en la comunidad, con múltiples fuentes capaces de emitir el virus a varias distancias.
Mito 4: «Si el número reproductivo básico, R0, no es tan grande como para el sarampión, entonces no puede ser aerosol”
El número reproductivo básico o R0 se define como el número medio de casos secundarios derivados de un único caso índice infectado en una población distribuida uniformemente y totalmente susceptible. El problema es que este R0 no está directamente relacionado con si una enfermedad se transmite o no por inhalación de aerosoles.
Diversos microbios pueden diseminarse por vía aerotransportada, pero no se transmiten necesariamente de persona a persona. Por ejemplo, los hantavirus y el Bacillus anthracis, causante del ántrax, tienen reservorios animales y ambos se adquieren por inhalación, pero no se transmiten de persona a persona. Tienen un R0=0 y, sin embargo, se consideran enfermedades transmitidas por el aire.
En el caso de los virus aéreos, como el sarampión y la varicela, la identificación precisa de los casos es relativamente sencilla porque estos virus causan una patología cutánea distintiva en el 99% de los infectados. Y pueden diagnosticarse sin pruebas de laboratorio. Las estimaciones del R0 son, por lo tanto, mucho más precisas. Dado que muchos casos de covid-19 son asintomáticos, el R0 es mucho más difícil de evaluar.
Cuando los pacientes presentan una ‘enfermedad similar a la gripe’, con síntomas leves o ninguno en absoluto, el alcance de cualquier brote y, en consecuencia, el número de casos secundarios, es mucho más difícil de determinar. Las personas no necesariamente sabrán que han estado expuestas o serán conscientes de su capacidad para transmitir la infección a otros. No se autoaislarán y no se contarán como posibles casos secundarios.
Esto hace que sea imposible el rastreo de contactos y el seguimiento de todos los involucrados en un evento de exposición específico, a menos que haya un registro muy detallado. Además, no se pueden excluir otros contactos que podrían haber contagiado desde una fuente diferente. Incluso en los casos en los que un único brote pueda asociarse a una fuente concreta, esa misma fuente puede haber propagado ya otros casos secundarios que no se puedan rastrear ni contabilizar. Puede haber una cantidad sustancial de transmisión presinstomática y además no todos los infectados son igualmente contagiosos.
Mito 5a: «Si es un aerosol, entonces las mascarillas no sirven»
Esta afirmación es falsa porque se presenta esencialmente como un escenario binario simplificado, es decir, las mascarillas funcionan (completamente) o no funcionan (en absoluto) contra virus en partículas respiratorias.
Varios estudios de laboratorio ya han demostrado que las mascarillas quirúrgicas y caseras son algo eficaces tanto para frenar las partículas exhaladas como para proteger de la inhalación de partículas ajenas. Contienen y reducen esas partículas hasta 3-4 veces (es decir, ∼67-75%), e incluso el 100% en el caso de coronavirus estacionales. Además, cuando un contagiado usa una mascarilla, el tamaño de su penacho exhalado también se reduce.
Las mascarillas quirúrgicas protegen al usuario al reducir la exposición a las gotas entrantes y aerosoles de infectados en un promedio de seis veces en función de su capacidad de filtración. Incluso las de tela caseras pueden reducir la exposición de las partículas entrantes hasta en 2-4 veces (es decir, ∼50-75%). La experiencia con el SARS-CoV-2 recomienda los modelos N95/FFP2/FFP3 para los sanitarios de primera línea. Para los que no pueden tolerar esas mascarillas durante largos períodos, las quirúrgicas ofrecen cierta protección pero no son tan efectivas.
Mito 5b: «Si el virus solo mide 100 nanómetros (0,1 µm), las mascarillas y los filtros no funcionan»
Hay dos malentendidos en este asunto. En primer lugar, hay una falta de comprensión de cómo funcionan realmente los filtros de alta eficiencia. No actúan como simples cedazos o coladores, sino que eliminan físicamente las partículas utilizando una combinación de impacto e interceptación (donde las partículas en movimiento más rápido golpean y se pegan a las fibras de la mascarilla a través de una colisión directa); tamización (donde las partículas en movimiento más lento tocan y se adhieren a las fibras de la mascarilla); y fuerzas electrostáticas (donde las partículas de carga opuesta y las fibras de la mascarilla se adhieren entre sí). Juntos, estos factores crean una «trampa de colisión dinámica» a medida que las partículas pasan a través de la red de canales de aire entre fibras a varias velocidades.
La eficiencia mínima de filtración se produce típicamente para partículas de unos 0,3 µm de diámetro. Aquellas más pequeñas se capturan con mayor eficiencia porque su movimiento browniano (que permite la difusión a nivel atómico) hace que colisionen con las fibras del filtro a una alta velocidad. Y las partículas más grandes que este diámetro se eliminan a través del impacto y la interceptación.
En segundo lugar, los virus no suelen ir ‘desnudos’ por el aire. Son expulsados en gotas que contienen agua, sal, proteínas y otros componentes de las secreciones respiratorias. Las gotas salivales y mucosas son mucho más grandes que el virus, y es el tamaño total el que determina cómo se mueven las gotas y aerosoles y son capturados por las mascarillas y los filtros.
Los filtros de aire de alta eficiencia (HEPA) pueden atrapar el 99,97 % o más de partículas de 0,3 µm (300 nanómetros) de diámetro. Las gotas salivales/mucosas exhaladas comienzan a partir de un tamaño de 0,5 µm y se eliminan por completo mediante filtros HEPA. De hecho, la filtración HEPA no es estrictamente necesaria en los sistemas de ventilación de la mayoría de los edificios comerciales, aunque sí en los hospitalarios. Los limpiadores de aire ‘portátiles’ que filtran el aire de una sala a través de filtros HEPA integrados son una opción para áreas no especializadas, como oficinas y aulas, aunque su rendimiento puede estar limitado por mezclas imperfectas, ruidos y corrientes.
Mito 6: «A menos que crezca en cultivos, no es infeccioso»
El cultivo viral es sorprendentemente difícil, una de las razones por las que el aislamiento de virus en cultivo celular es mucho menos sensible que su detección por métodos moleculares. Esto se debe en parte a que se necesita más de un virus para infectar con éxito un cultivo celular. Por ejemplo, con la gripe la cantidad de virus necesaria para infectar el 50% de una monocapa celular in vitro es de unas 300 copias de su genoma.
Esta diferencia de sensibilidad se ve agravada por las técnicas de muestreo de aire disponibles actualmente. La mayoría de los estudios utilizan sistemas de alta velocidad que succionan cualquier virus en el aire a un medio de cultivo de líquidos burbujeantes. Sin embargo, estos dispositivos de muestreo de aire generan altas fuerzas de cizallamientoque pueden dañar las proteínas virales de la superficie y evitar que crezcan en el cultivo.
Por el contrario, las velocidades naturales de exhalación humana y flujo de inhalación son mucho más lentas, lo que las hace menos propensas a deteriorar los virus. Es decir, nuestras tecnologías de muestreo de aire no replican con precisión los mecanismos que conducen a la infección respiratoria humana por inhalación.
Como consecuencia, la falta de detección de virus viables en muestras de aire no prueba necesariamente la ausencia de virus vivos en muestras donde el ARN viral se detecta por métodos moleculares. Encontrar ARN viral en muestras de aire debe interpretarse como presencia de virus vivos, según el principio de precaución, que siempre debe reforzar el control eficaz de la infección. Para el SARS-CoV-2, dos grupos de investigación han demostrado la presencia de virus infecciosos en muestras de aerosoles de salas de pacientes. Por las razones antes mencionadas, es muy probable que estos estudios subestimen la cantidad de aerosoles viables e inhalables.
En resumen: no hay que despreciar la capacidad infecciosa de la exposición a pequeñas partículas en el aire, que puede ser igual de probable que la transmisión más aceptada a través de gotas respiratorias más grandes o del contacto directo con personas infectadas o superficies contaminadas. Las pruebas que se van acumulando refuerzan la necesidad de equipos de protección personal, de ventilación suficiente y eficaz, de control de hacinamientos en atención sanitaria, transportes e interiores, y de estrategias de higiene, desinfección, filtración y limpieza ambiental
Dentro de cada célula del cuerpo humano se encuentra material genético conocido como adn aquí se contienen las instrucciones genéticas para el desarrollo y funcionamiento de todos los organismos vivos y uno que otro virus está se encarga de transmitir rasgos hereditarios a futuras generaciones y por ello es un componente esencial para la vida gracias a esto cada célula sabe qué tiene que hacer para formar tejidos manejar energía del cuerpo construir defensas y sobre todo mantenernos vivos los procesos que realiza cada célula parecen precisos pero no son perfectos ya que el adn que contiene cada célula puede llegar a ser alterado por un sinfín de razones como enfermedades lesiones o sustancias químicas lo cual puede producir errores que generan cambios estos cambios nos conocemos comúnmente como mutaciones, una mutación es una alteración o cambio en la información genética de un ser vivo en su secuencia de doble cadena unidas entre sí formando combinaciones esenciales y una alteración en esta secuencia puede variar desde simples cambios estéticos del cuerpo humano hasta enfermedades genéticas las cuales son muy difíciles de tratar y eliminar debido a que están situadas en el código genético de cada célula una manera de eliminar estas enfermedades que surgen de errores en el adn es modificando la secuencia y aquí es donde las cosas se ponen interesantes porque esto significa que prácticamente se debe crear un editor de texto suena a ciencia ficción pero esto ya es una realidad todo esto es posible gracias a crist que en español significa
Cuando una bacteria es atacada por un virus este debe defenderse a toda costa si sobrevive guardo un pedazo del adn del virus que él atacó y lo agrega a su genoma solo que al hacer esto genera una conexión para aislarlo he aquí la repetición una vez teniendo esto la bacteria genera inmunidad contra ese virus y este es información que hereda a futuras generaciones de esta colección de adn se genera a rn el cual es guardado en una proteína llamada cast 9 ésta es la que se encarga de buscar identificar y desactivar el virus en caso de que vuelva a atacar ahora el adn contiene secuencias repetidas que se encuentran tanto en genomas eucariotas como en procariotas estas secuencias son conocidas erróneamente como adn basura debido a que no codifican para proteínas estudios realizados por biólogos han demostrado que estas secuencias tienen varias aplicaciones entre ellas reparar regular y marcar partes del adn esto con el fin de modificar el adn para combatir enfermedades repararse o simplemente controlar el proceso de corte y empalme del adn estas secuencias repetidas pueden ser utilizadas como marcadores e interruptores para cast 9 esto significa que se puede programar una proteína casi 9 con arn para modificar eliminar o agregar partes del adn y gracias a las secuencias repetidas al momento de editar el adn se evita cortar demás la secuencia lo cual aumenta la precisión y evita los problemas que se dan al momento de modificar el adn con químicos o radiación ósea dejamos de editar aleatoriamente el adn y lo convertimos en algo más preciso la manipulación genética es algo que se ha hecho desde que empezamos a controlar la selección natural en granjas y laboratorios para crear animales con más carnes plantas más resistentes e incluso curarnos de enfermedades crisis pero lo que hace es abrir todo un panorama de aplicaciones de mejoras para cosas que ya hacemos pero como se lo pueden imaginar tiene serias implicaciones morales y éticas
El científico estadounidense James Wason y el británico Francis Crick culminaron su descubrimiento de la estructura molecular del ADN, en forma de doble hélice. Ese hallazgo revolucionó entonces la ciencia, al permitir entender cómo funciona la molécula portadora del programa genético de los organismos vivos. Ahora, esto tiene implicaciones aún más profundas gracias a la tecnología de edición de genes CRISPR —las siglas en inglés de repeticiones palindrómicas cortas agrupadas y regularmente espaciadas—, que permite a los científicos cortar y alterar con precisión el ADN de cualquier célula.
Aunque CRISPR —también conocido como “tijera molecular”— aún no ha curado enfermedades ni ha acabado con el hambre en el mundo, ya se está utilizando de algunas maneras sorprendentes.
Convertir cerdos en donantes de órganos
Durante décadas, la mejor solución que han concebido los científicos para reducir la lista de las miles de personas que esperan recibir un trasplante de órganos en todo el mundo ha sido utilizar órganos de animales en humanos. Por ejemplo, el primer trasplante de corazón se realizó en 1964, cuando el órgano de un chimpancé fue implantado en un humano, que falleció dos horas después de la cirugía.
Además de que el cuerpo humano rechaza tejidos extraños, otro riesgo de esa alternativa es la posibilidad de que las infecciones de los animales puedan transmitirse a los receptores humanos. Pero la empresa eGenesis, que nació en el laboratorio del genetista George Church de la Universidad de Harvard, cree que el CRISPR puede resolver o eliminar estos obstáculos.
La empresa eGenesis ha utilizado la edición genética para eliminar una familia de virus que se encuentran en el ADN de los cerdos. Crédito: eGenesis
La compañía también está experimentando con CRISPR para modificar los genes relacionados con el sistema inmunológico y evitar que el cuerpo humano rechace los órganos de donantes. Sin embargo, los científicos advierten que todavía quedan algunos años para que se pueda hacer un ensayo clínico de trasplantes humanos con órganos producidos en cerdos genéticamente modificados.
Alternativas a la insulina
Las personas con diabetes tipo 2 (resistente a la insulina) podrían tener una opción para sustituir las inyecciones con un injerto de piel. Se trataría de un injerto que contiene una versión modificada por CRISPR de una proteína que ayuda a la insulina a regular los niveles de glucosa en la sangre. Investigadores de la Universidad de Chicago están utilizando CRISPR para para alterar el gen GLP-1, responsable de la codificación de la hormona péptido 1, que provoca la liberación de insulina y luego ayuda a eliminar el exceso de glucosa de la sangre.
Usando CRISPR, los científicos han comprobado que el gen GLP-1 podría modificarse para que sus efectos de regulación tengan larga duración. Cerca del 80% de los injertos de piel que se aplicaron en ratones liberaron con éxito la hormona editada en la sangre, regulando los niveles de glucosa durante cuatro meses y revirtiendo la resistencia a la insulina y el aumento de peso en los pacientes.
Investigadores de la Universidad de Chicago están utilizando CRISPR para para alterar el gen GLP-1. Crédito: University of Chicago
Los tratamientos en humanos tardarán tiempo en desarrollarse, pero la buena noticia es que los científicos ya pueden hacer crecer el tejido de la piel muy fácilmente en el laboratorio utilizando células madre. La previsión es que esa técnica pueda tratar también enfermedades como la hemofilia (cuando el cuerpo no puede hacer los coágulos de sangre de manera adecuada).
Acabar con enfermedades endémicas
Las enfermedades transmitidas por mosquitos, especialmente la malaria, matan a más de 400.000 personas cada año en todo el mundo. Para reducir esa cifra, algunos científicos proponen utilizar una tecnología llamada unidad genética. Se trata de una herramienta de ingeniería genética diseñada para diseminar ciertos genes a través de una especie. Y aunque no es una idea nueva, estas unidades de genes están más cerca de ser realidad gracias al CRISPR.
En un artículo publicado en septiembre de 2018, los investigadores del Imperial College de Londres mostraron que una unidad genética realizada con CRISPR podría suprimir una población de Anopheles gambiae, el tipo de mosquito que transmite la malaria en el África subsahariana. Los investigadores utilizaron el “corta y pega” genético para atacar el gen Doublesex, responsable por el desarrollo femenino. Cuando los mosquitos hembra heredaron dos copias de este gen modificado, no pudieron picar ni poner huevos.
Una unidad genética realizada con CRISPR podría suprimir una población de Anopheles gambiae, el mosquito que transmite la malaria. Crédito: James D. Gathany
Los investigadores pusieron esos mosquitos en jaulas y encontraron que eran autodestructivos para su especie en su entorno cercano: después de ocho generaciones, ya no quedaban hembras normales para reproducirse y la población se extinguió.
Ese tipo de experimentos no han sido realizados fuera de los laboratorios todavía —existe la posibilidad de que las alteraciones genéticas diseñadas para impactar las poblaciones puedan mutar y transmitir rasgos ventajosos a las demás generaciones—, pero ese estudio comprobó que se transmitió la modificación genética casi el 100% de las veces, evitando la resistencia.
La herramienta de edición de genes interrumpió con éxito el gen DFR-B, que es responsable del color de los tallos, las hojas y los pétalos, cambiando así el color violeta característico de la flor al blanco.
EL Dr Mujica, hizo un descubrimiento extraordinario al señalar y cortar una secuencia de ADN patologico, y poder intercalar otro ADN normal, en levaduras de la Sal en Alicante
Pero recientemente , se ha simplificado la técnica, que en vez de recortar la secuencia patológica de ADN, añade al genoma del paciente una copia en buen estado del gen dañado, para que recupere su función normal y la enfermedad remita.
CRISPR-Cas9 actúa como unas tijeras que cortan la doble hélice del ADN, lo que a veces puede desencadenar cambios no deseados en las letras o bases (A, T, G, C) que escriben el genoma. Si se consiguen eliminar esos “efectos secundarios”. La técnica, cambiaria la estrategia de “cortar y pegar” por un sistema de edición de textos -“buscar y reemplazar”- de tal precisión, que en teoría podría corregir alrededor del 89% de las variantes genéticas humanas asociadas con enfermedades. “Si CRISPR son las tijeras, los editores de bases serían el lápiz: en lugar de cortar la doble hélice, convierten una letra del ADN en otra, sin llegar a romper la doble cadena, lo que permite corregir los principales tipos de mutaciones de forma eficiente, pero no todas. El editor ‘prime’ supondría el sistema de ‘búsqueda y sustitución’ de un procesador de texto; permite realizar directamente mutaciones puntuales específicas, inserciones y eliminaciones de una sola letra y combinaciones de estas, también sin tener que romper la doble cadena”, La tecnología de edición genética CRISPR-Cas9 ha revolucionado la investigación biológica y médica, al proporcionar la herramienta más sencilla con la que editar el ADN. Su potencial es enorme: desde corregir mutaciones asociadas a enfermedades a obtener plantas más resistentes para el cultivo.
La gran innovación de esta técnica prime consiste en la fusión de la proteína Cas9 –que es la encargada de cortar el ADN en el sistema de edición clásico– con una enzima de transcriptasa inversa –molécula que genera ADN a partir de ARN- y la modificación de la guía de ARN para que, a la vez que localiza el sitio que se quiere editar, actúe de molde para corregira la mutación. De esta forma, se evita la rotura de doble cadena. Los científicos han probado la técnica con más de 175 ediciones genéticas en células humanas, incluida la corrección del error que causa la anemia de células falciformes y la enfermedad de Tay Sachs, una patología por depósito lisosomal que afecta al sistema nervioso central. Según exponen en el artículo, la técnica es muy eficiente y produce menos “efectos secundarios” que la clásica CRISPR-Cas9. Pero esta investigación es un “cambio revolucionario” que se produce en la técnica del CRISPR. Este trabajo ofrece , un método robusto de corrección de alelos patogénicos Y abre un gran número de posibilidades biotecnológicas. Permite corregir pequeños alelos con un nivel de certidumbre más alto que métodos anteriores y en un rango de condiciones muy grande (incluyendo células que se dividen poco). También parece que tiene un nivel bajo de off-target (ediciones fuera de diana)”. Güell considera que si bien el CRISPR-Cas9 clásico funciona muy bien “para romper o inactivar genes”, el editor prime parece “bastante superior para corregir” errores genéticos. Y, además, “potencialmente es más seguro”, al evitar la rotura de doble cadena de ADN.
Los resultados de una nueva tecnología llamada Uni-large, desarrollada para modificar el genoma para hacer frente a diversas enfermedades de manera más segura y eficiente que otras soluciones, están ya en fase de revisión para su publicación, ha explicado a Diario Médico Marc Güell, investigador principal de este proyecto en el Grupo de Investigación en Biología Sintética Traslacional del Departamento de Ciencias Experimentales y de la Salud (DCEXS) de la Universidad Pompeu Fabra (UPF) de Barcelona.
Uni-large está pensada para tratar las enfermedades de origen genético y algunos tipos de cáncer derivados del mal funcionamiento de un gen (como la leucemia, por ejemplo). Según información de la UPF, consiste en añadir al genoma del paciente una copia en buen estado del gen dañado, para que recupere su función normal y la enfermedad remita. La universidad ha patentado esta tecnología y previsto su protección a la par que ha creado una empresa, Integra Therapeutics, que desarrollará y comercializará los programas terapéuticos vinculados a ella, ha precisado Güell.
Inicialmente el equipo está probando su utilidad en el tratamiento de la distrofia muscular congénita tipo 1A (MDC1A), una enfermedad hereditaria para la que no existe ninguna terapia efectiva. Se trata de la distrofia muscular congénita más frecuente y provoca debilidad progresiva y pérdida de la masa muscular.
Entre las ventajas de Uni-large destaca su universalidad puesto que otras técnicas de edición genética, como la CRISPR, pueden corregir mutaciones individuales de cada paciente (medicina personalizada), mientras que la nueva tecnología sería útil con todos los pacientes de una determinada enfermedad. Además, con CRISPR hay que hacer un corte en el genoma para reparar la mutación, lo que puede provocar accidentes como la pérdida o la rotura de cromosomas, mientras que con Uni-large no se corta, sólo se pega un gen, lo que evita riesgos. Además, el método es especialmente útil con enfermedades como la MDC1A donde el gen implicado es muy grande y otras tecnologías no permiten reparaciones eficientes.
Güell recalca especialmente que Uni-large es de la misma familia de la tecnología CRISPR, pero la suya está pensada para poner un gen entero; es decir, permite escalar “muy bien”, lo cual la hace universal.
Inmediatamente ha surgido una modificación
Explica que hace años, tras una estancia en Estados Unidos, empezó a trabajar en la idea de combinar la precisión de las tecnologías modernas pero sin perder de vista la clásica. Probaron con células en cultivos y se fueron realizando ajustes hasta llegar a un modelo de ratón con distrofia muscular donde ya se ha visto que funciona “bastante bien”.
Fue Francis Mojica, quien realizó la primeras contribuciones que describían las secuencias repetidas CRISPR en arqueas y su papel en los mecanismos de inmunidad de las células procariotas. Sus descubrimientos cristalizaron más tarde en el desarrollo de la tecnología CRISPR-Cas.
Referencia Sonia Moreno Biología Molecular Carmen Fernández. Barcelona 21 octubre, 2019
Marc Güell, en el edificio del PRBB de Barcelona. 16/01/2021 –
Vacunas de ARN mensajero, son aquellas que en las que se emplea ácido ribonucleico para lograr el desarrollo de una respuesta inmune. Se diferencian de las vacunas tradicionales en que no se administran agentes vivos atenuados ni fragmentos del mismo, por lo que no existe el peligro de provocar la enfermedad que se pretende prevenir. Para fabricarlas es preciso encontrar las secuencias de ADN que codifican antígenos esenciales del agente infeccioso y después transcribirlo para obtener el ARN correspondiente, el cual se usará como vacuna. Aunque existen diferentes tipos de ARN, en las vacunas se utiliza ARN mensajero. Una vez administrada, parte del ARN puede degradarse por acción de las ARNasas, pero la porción que entra en las células genera péptidos similares a los del agente patógeno, lo que provoca una respuesta inmune que protege de la infección. 1 2 3 4 5
Este proceder es revolucionario y puede ser aplicado y varias enfermedades autoinmunes.
Posiblemente no fue puesto en marcha, hasta que se conoció mejor, la producción desde el ARN, de anticuerpos específicos, sin peligrosidad de enfermedad, ni perdida de inmunidad
Liposoma cargado de ARN mensajero.
Microesferas de lípidos (liposomas) cargadas de ARNm penetran en la célula por un proceso de endocitosis.
Esquema general del proceso de traducción genética mediante el cual se sintetiza una proteína a partir del ARN mensajero (mRNA).
Las vacunas tradicionales contienen el agente infeccioso inactivado o fragmentos del mismo que al introducirse en el cuerpo provocan una respuesta inmune por parte del organismo, el cual de esta forma responde con gran rapidez y eficacia cuando sufre una infección verdadera por el microorganismo específico para el que está diseñada la vacuna. Sin embargo las vacunas de ARN consisten en una secuencia de ácido nucleico que introduce en la célula el código para que la maquinaria celular fabrique la proteína extraña del agente infeccioso, la cual posteriormente es presentada en la membrana celular y reconocida por el sistema inmune, que genera inmunidad contra el mismo; por lo tanto puede decirse que no introduce el antígeno, sino las instrucciones para fabricarlo. 6
El ARN mensajero o ARNm es el ácido ribonucleico que transfiere el código genético desde el ADN a los ribosomas en el citoplasma de una célula. Actúa por tanto como plantilla o patrón para la síntesis de una proteína. Se trata de un ácido nucleico de cadena única (monocatenario), a diferencia del ADN, que tiene dos cadenas enlazadas (bicatenario). Las vacunas de ARN mensajero están formadas por cadenas de esta molécula que codifican un antígeno específico de un patógeno. Cuando el ARNm entra en la célula, el ribosoma sintetiza la proteína codificada que corresponde a un antígeno del patógeno, el cual posteriormente se presenta en la superficie de la célula, donde es reconocido por las células del sistema inmune, generando inmunidad.
Para evitar la rápida degradación de la molécula antes de entrar a la célula, se utilizan varias estrategias, una de ellas emplea microesferas de lípidos (liposomas) en cuyo interior se encuentra el ARN, que de esta forma entra en la célula con facilidad por un proceso de endocitosis. La idea de encapsular ARNm en nanopartículas lipídicas ha resultado atractiva por varias razones. El recubrimiento de lípidos proporciona una capa de protección que evita la rápida degradación, lo que hace posible un proceso de traducción genética para formación de proteínas más eficiente. Además, la capa externa de lípidos puede modificarse, lo que permite que se una a las células deseadas a través de interacciones de ligandos. Las nanopartículas pueden administrarse al organismo a través de diferentes rutas, por ejemplo por vía intravenosa o por inyección intramuscular.
Determinadas vacunas utilizan ARN autoampliflicable (replicón)Nota 1, es decir, el ARN introducido se multiplica por sí mismo en el interior de la célula, lo que hace que se genere una cantidad muy superior del antígeno contra el que se pretende crear inmunidad. Esta técnica no pueden producir agentes infecciosos activos porque se ha eliminado el gen de la proteína estructural del virus y este no puede formarse completo ni propagarse a las células adyacentes.789
Una de las particularidades de las vacunas de ARN es que desencadenan la respuesta inmune mediante varios mecanismos. Estimulan la formación de anticuerpos y reclutan linfocitos T citotóxicos mediante la unión de la proteína virica producida en los ribosomas al complejo mayor de histocompatibilidad tipo I (MHC). Este doble mecanismo no tiene lugar con otros tipos de vacunas.
Mecanismo de acción de las vacunas de ARN e interacción con el complejo mayor de histocompatibilidad (MHC)
Las vacunas de ARN son menos estables que otros tipos de vacunas y pueden ser degradadas fácilmente por el calor. Por ello deben conservarse congeladas o a temperaturas muy bajas, lo que representa un inconveniente para el proceso de distribución. 2
Las ventajes potenciales de las vacunas de ARN son:
Seguridad. No se inoculan microorganismos vivos ni atenuados, por lo que no existe la posibilidad de provocar una infección.3
El ARN no se integra en el genoma del hospedador, que está formado por ADN, por lo que no existe la posibilidad de alterar el genoma. 3
El ARN se degrada con relativa rapidez, lo que podría evitar la aparición de efectos secundarios a largo plazo. 3
El proceso de producción puede ser rápido y más estandarizado que en las vacunas tradicionales, lo que facilitaría una rápida respuesta ante la aparición de nuevos agentes infecciosos.3
Cáncer Las vacunas de ARN contra el cáncer no son preventivas, están diseñadas para el tratamiento de personas que ya están diagnósticadas de esta enfermedad e intentan potenciar el sistema inmunológico para que destruya las células malignas del tumor. 25 26
Los primeros estudios sobre la eficacia de vacunas de ARNm fueron realizados por Woff en 1990. Posteriormente se desarrollaron dos formas: vacunas de ARNm convencional y vacunas de ARNm autorreplicativo. Los trabajos iniciales no alcanzaron resultados prácticos por la fragilidad de la molécula de ARN y su inactivación por endonucleasas, sin embargo con el tiempo se han desarrollado métodos que aumentan la estabilidad del ARNm y permiten su producción sintética en el laboratorio a partir de plásmidos de ADN, mediante una transcripción enzimática y ARN polimerasa, sin que sean precisos cultivos celulares.27
La actitud de nuestros investigadores para controlar la epidemia de coronavirus es algo genial y admirable. Una Legión de investigadores se han volcado para ayudar al mundo y lo están consiguiendo .
Describo a continuación los distintos tipos de vacunas
La vacuna de la Universidad de Oxford y AstraZeneca, conocida como AZD1222, cuya comercialización acaba de aprobar Reino Unido, utiliza un vector viral no replicante (adenovirus de chimpancé) que contiene genes que codifican la proteína S del coronavirus SARS-CoV-2. Al igual que las vacunas de ARNm -la de Pfizer+BioNTech y la de Moderna– se basan en una tecnología novedosa; la única vacuna con vector de adenovirus aprobada se dirige a la enfermedad del Ébola y es de Janssen, que también está ensayando una vacuna por esta vía para el coronavirus.
Los datos que avalan la seguridad y eficacia de la vacuna recién aprobada se basan en cuatro ensayos controlados llevados a cabo en 23.745 participantes en el Reino Unido, Brasil y Sudáfrica. Los resultados del análisis intermedio (11.636 personas) de la fase III se publicaron en The Lancet.
La interrupción del ensayo debido a un caso de mielitis tranversa (que finalmente se localizó en el grupo que no había recibido la vacuna) fue el primer revés que se vivió en esta carrera científica bajo los focos. Salvo la mielitis y una anemia hemolítica (también en el grupo control), que se han resuelto, no se han comunicado efectos graves en el ensayo. A diferencia de las vacunas de Pfizer y de Moderna, no se ha informado ningún caso de parálisis de Bell.
La reactogenicidad es la esperable en una vacuna, todos son síntomas leves, y quizá se presenten con algo más de frecuencia que en la vacunad de ARNm. Los estudios indican que hasta un 60% de los sujetos refirieron sensibilidad en el punto de la inyección; una de cada dos personas refieren fatiga, dolor en el lugar del pinchazo y dolor de cabeza, y más de un 30% tienen fiebre.
Los estudios indican con los datos combinados de estos estudios una eficacia del 70,4% en la prevención de la enfermedad, pero un error en el desarrollo de uno de los ensayos reveló que no todos los participantes habían recibido las dos dosis completas estipuladas. De hecho, en aquellas personas que recibieron media dosis primero y una segunda dosis después se observó una eficacia superior, del 90%, mientras que entre los que habían recibido las dos dosis completas, la eficacia en el ensayo se estableció en un 62,1%.
Hay algunas hipótesis que explicarían por qué media dosis y una entera obtendrían (si se confirma que es así) más eficacia que las dos dosis completas, y que tiene que ver con la potencial respuesta inmune provocada por el vector de la vacuna, el adenovirus, como sintetiza, Jaime Pérez Martín vocal de la Asociación Española de Vacunología. “No obstante, los datos que hay publicados no permiten afirmar claramente que sea más protectora una pauta que otra”.
El dato de eficacia contrasta con el aportado por las dos vacunas de ARN mensajero que desarrollaron Pfizer y BioNTech, por un lado, y Moderna, por otro. La primera ya está aprobada por la FDA y la EMA, y ha comenzado a administrarse en España, mientras que la de Moderna está aún a la espera del visto bueno de la agencia europea, que previsiblemente se producirá la semana que viene, tras recibir el respaldo de la FDA hace unos días. Los estudios con estas vacunas indican que frente a placebo, la inmunización protege de enfermedad en más del 90%, pero, desde la Sociedad Española Medicina Preventiva, Salud Pública e Higiene (Sempsph) hacen hincapié en que los estudios no han comparado un tipo de vacuna con otras.
“Los datos de eficacia que se aportan –un 70% en el caso de la vacuna de AstraZeneca, y un 90% en el de Pfizer- se han obtenido frente a placebo, no son equiparables entre sí. Esto solo podría saberse si se hiciera un ensayo clínico de una frente a la otra”, enfatizan fuentes de la sociedad científica.
La aprobación británica recoge que la administración será de dos dosis de 0,5 ml que se administrarán con un intervalo de entre cuatro y doce semanas. Una característica de esta vacuna, a diferencia de la de ARNm recientemente aprobada en la Unión Europea, es que cuanto mayor sea el tiempo de separación entre la primera y la segunda dosis, mayor es la generación de anticuerpos. Aún hay que determinar si ese aumento de la separación se corresponde con una mayor protección. No obstante, en este contexto pandémico puede parecer poco oportuno esperar tres meses para obtener la inmunidad idónea.
La vacuna se indica a partir de los 18 años -dos más que la indicación de la de Pfizer y el mismo umbral mínimo establecido para la de Moderna- y su eficacia en mayores de 65 años no está tan medida como en otros estudios, si bien casi un 6% de los participantes de los ensayos se encuentran en esta franja de edad, la baja incidencia de infecciones impidió tener datos robustos de eficacia. No obstante, en este grupo poblacional se constató, a través de la medición de anticuerpos, una respuesta adecuada. El perfil de seguridad fue similar con respecto al grupo de 18 a 64 años.
Con todo, la ficha técnica de la vacuna dice que “los datos de eficacia y seguridad son limitados en individuos mayores de 65 años”. Para Pérez Martín parece plausible con esta información que el objetivo de esta vacuna sea una población más joven, si bien “es una estrategia que deben definir los expertos en salud pública británicos. No olvidemos que hay que vacunar a mucha a gente».
Además de los ensayos con los que ha logrado la aprobación británica, la compañía tiene en marcha también un ensayo de combinación de su vacuna con la rusa, conocida como Sputnik V (Ad26), basada también en un vector viral, aunque en este caso humano, con el objetivo de averiguar si mejora así la eficacia de la de AstraZeneca.
Una ventaja diferenciadora de esta vacuna frente, en concreto frente a la de Pfizer+BioNTech, son las condiciones de refrigeración necesarias para su manejo. Así, esta última requiere una temperatura de conservación de unos -75°C±15°C, aunque la multinacional estadounidense ha desarrollado un sistema para facilitar en gran medida la logística de distribución y almacenamiento hasta el punto final de administración de su vacuna gracias a una sofisticada infraestructura, con torre de control incluida, para el seguimiento permanente de los viales. La vacuna aprobada hoy de AstraZeneca -y la de Moderna-, por su parte, pueden mantenerse a una temperatura entre 2º y 8º, con lo que pueden ser almacenadas durante al menos seis meses, transportadas y manipuladas en condiciones de refregeración habituales en la vacunación y, por tanto, administradas con facilidad en los centros sanitarios.
Junto a esta característica favorable, y para lograr la mayor distribución posible de la vacuna, AstraZeneca «ha aprovechado su propia capacidad industrial y se ha asociado con más de 20 colaboradores para la distribución de la vacuna en más de 15 países apoyados por más de 20 centros de analítica para establecer cadenas de suministro en paralelo en un tiempo récord y garantizar así el acceso global a la vacuna, con el objetivo de alcanzar los 3.000 millones de dosis, según explica Per Alfredsson, vicepresidente de Operaciones Biológicas de la compañía.
La vacuna de Janssen una dosis única
‘The New England’ publica resultados de un ensayo en fase 2 con la vacuna Ad26.COV2.S, una de las vacunas que podría administrarse en una sola dosis.
La vacuna de Janssen se basa en un vector viral (adenovirus humano) sin capacidad de replicación.
Sonia Moreno
Jue, 14/01/2021 – 08:00
El análisis intermedio del ensayo en fase 1-2a de la candidata vacunal Ad26SARS-Cov-2 que desarrolla Janssen, filial europea de Johnson & Johnson, revela un buen perfil de seguridad y reactogenidad, así como de inmunogenicidad en un esquema de una dosis y de dos dosis, según acaba de publicar The New England Journal of Medicine.
Meses antes de que Araceli Rosario Hidalgo se convirtiera en la primera española en recibir una vacuna autorizada contra la covid, en España la candidata vacunal de Janssen era la primera inmunización que se administraba a los voluntarios en el contexto de un ensayo, que posteriormente se ha continuado a la fase 3 en varios hospitales de todo el país.
Esta inmunización se basa en un vector viral sin capacidad de replicación –adenovirus humano a diferencia del de chimpancé de la vacuna de Oxford y AstraZeneca- y utiliza la misma plataforma y tecnología que la vacuna de Janssen aprobada para la prevención del Ébola.
Los resultados que recoge ahora The New England se corresponden a un ensayo realizado en doce centros de Bélgica y Estados Unidos, sobre 805 participantes repartidos en un grupo de entre 18 y 55 años, y otro grupo específico de 65 años o más. En el estudio se han recabado datos frente a placebo de la administración de una dosis baja o una dosis alta en un esquema de una sola dosis o de dos dosis.
Como es de esperar en esta fase de ensayo, los criterios de valoración principales fueron la seguridad y la reactogenicidad de cada programa de dosis.
Tras la administración de la primera dosis de vacuna, los eventos adversos más frecuentes fueron fatiga, dolor de cabeza, mialgia y dolor en el lugar de la inyección. El evento adverso sistémico más frecuente fue la fiebre. Los efectos adversos sistémicos fueron menos frecuentes entre los mayores que en los individuos más jóvenes, y en los que recibieron la dosis baja de la vacuna que en los que recibieron la dosis alta. Al contrario de lo que se ha observado con las vacunas de ARN mensajero, la reactogenicidad fue menor después de la segunda dosis.
El trabajo, que ha dirigido la viróloga holandesa Hanneke Schuitemaker, de Janssen, está ahora recopilando datos a largo plazo para comparar el régimen de dosis única con dos dosis, por lo que aún no pueden extraerse conclusiones fehacientes al cotejar ambos regímenes. No obstante, en esta publicación se informa de que una dosis única de Ad26.COV2.S provocó una respuesta humoral fuerte en la mayoría de los que la recibieron, con la presencia de unión a la proteína coronavírica S y anticuerpos neutralizantes en más del 90% de los participantes, independientemente del grupo de edad o de la dosis.
Los autores escriben que “durante 71 días de seguimiento tras la primera dosis, los títulos de anticuerpos aumentaron y se estabilizaron aún más, lo que sugiere la durabilidad de la respuesta inmune provocada por Ad26.COV2.S”.
“Una vacuna eficaz de dosis única para la covid-19 tiene obvias ventajas logísticas sobre una vacuna de dos dosis, especialmente durante una pandemia”, escriben los autores. “Observamos que entre los participantes de 18 y 55 años, una segunda dosis de vacuna el día 57 aumentó aún más el título de anticuerpos, un hallazgo que también estaba en línea con nuestras observaciones recientes en primates no humanos”.
El estudio está analizando ahora si una segunda dosis proporciona un beneficio adicional para mejorar la eficacia o la durabilidad, especialmente en personas de edad avanzada en quienes la respuesta inmune después de la primera dosis tendió a ser moderadamente más baja que la de los participantes más jóvenes.
Para los investigadores los hallazgos presentados en este estudio, junto con los resultados de los estudios preclínicos, respaldan la decisión de avanzar en dos ensayos de fase 3 para evaluar la eficacia de un régimen de dosis única o de dos dosis de la dosis más baja de la vacuna.
La compañía confía en poder anunciar los primeros datos de la fase 3 de su vacuna candidata administrada en dosis única a fines de enero de 2021. Si se demuestra que es eficaz con un buen perfil de seguridad, Janssen espera presentar una solicitud de autorización de uso de emergencia ante la FDA estadounidense poco después.
Gracias a todos estos investigadores
SOBRE LAS VACUNAS CONTRA CORONAVIRUS SARS-COV-2.
ASTRAZENECA, PFIZER ,MODERNA Y JANSSEN
La actitud de nuestros investigadores para controlar la epidemia de coronavirus es algo genial y admirable. Una Legión de investigadores se han volcado para ayudar al mundo y lo están consiguiendo .
Describo a continuación los distintos tipos de vacunas que se lo siento coche calle perpendicular no tiene día cuenta marca están utilizando en España crees que sea Patricia mayo
La vacuna de la Universidad de Oxford y AstraZeneca, conocida como AZD1222, cuya comercialización acaba de aprobar Reino Unido, utiliza un vector viral no replicante (adenovirus de chimpancé) que contiene genes que codifican la proteína S del coronavirus SARS-CoV-2. Al igual que las vacunas de ARNm -la de Pfizer+BioNTech y la de Moderna– se basan en una tecnología novedosa; la única vacuna con vector de adenovirus aprobada se dirige a la enfermedad del Ébola y es de Janssen, que también está ensayando una vacuna por esta vía para el coronavirus.
Los datos que avalan la seguridad y eficacia de la vacuna recién aprobada se basan en cuatro ensayos controlados llevados a cabo en 23.745 participantes en el Reino Unido, Brasil y Sudáfrica. Los resultados del análisis intermedio (11.636 personas) de la fase III se publicaron en The Lancet.
La interrupción del ensayo debido a un caso de mielitis tranversa (que finalmente se localizó en el grupo que no había recibido la vacuna) fue el primer revés que se vivió en esta carrera científica bajo los focos. Salvo la mielitis y una anemia hemolítica (también en el grupo control), que se han resuelto, no se han comunicado efectos graves en el ensayo. A diferencia de las vacunas de Pfizer y de Moderna, no se ha informado ningún caso de parálisis de Bell.
La reactogenicidad es la esperable en una vacuna, todos son síntomas leves, y quizá se presenten con algo más de frecuencia que en la vacunad de ARNm. Los estudios indican que hasta un 60% de los sujetos refirieron sensibilidad en el punto de la inyección; una de cada dos personas refieren fatiga, dolor en el lugar del pinchazo y dolor de cabeza, y más de un 30% tienen fiebre.
Los estudios indican con los datos combinados de estos estudios una eficacia del 70,4% en la prevención de la enfermedad, pero un error en el desarrollo de uno de los ensayos reveló que no todos los participantes habían recibido las dos dosis completas estipuladas. De hecho, en aquellas personas que recibieron media dosis primero y una segunda dosis después se observó una eficacia superior, del 90%, mientras que entre los que habían recibido las dos dosis completas, la eficacia en el ensayo se estableció en un 62,1%.
Hay algunas hipótesis que explicarían por qué media dosis y una entera obtendrían (si se confirma que es así) más eficacia que las dos dosis completas, y que tiene que ver con la potencial respuesta inmune provocada por el vector de la vacuna, el adenovirus, como sintetiza, Jaime Pérez Martín vocal de la Asociación Española de Vacunología. “No obstante, los datos que hay publicados no permiten afirmar claramente que sea más protectora una pauta que otra”.
El dato de eficacia contrasta con el aportado por las dos vacunas de ARN mensajero que desarrollaron Pfizer y BioNTech, por un lado, y Moderna, por otro. La primera ya está aprobada por la FDA y la EMA, y ha comenzado a administrarse en España, mientras que la de Moderna está aún a la espera del visto bueno de la agencia europea, que previsiblemente se producirá la semana que viene, tras recibir el respaldo de la FDA hace unos días. Los estudios con estas vacunas indican que frente a placebo, la inmunización protege de enfermedad en más del 90%, pero, desde la Sociedad Española Medicina Preventiva, Salud Pública e Higiene (Sempsph) hacen hincapié en que los estudios no han comparado un tipo de vacuna con otras.
“Los datos de eficacia que se aportan –un 70% en el caso de la vacuna de AstraZeneca, y un 90% en el de Pfizer- se han obtenido frente a placebo, no son equiparables entre sí. Esto solo podría saberse si se hiciera un ensayo clínico de una frente a la otra”, enfatizan fuentes de la sociedad científica.
La aprobación británica recoge que la administración será de dos dosis de 0,5 ml que se administrarán con un intervalo de entre cuatro y doce semanas. Una característica de esta vacuna, a diferencia de la de ARNm recientemente aprobada en la Unión Europea, es que cuanto mayor sea el tiempo de separación entre la primera y la segunda dosis, mayor es la generación de anticuerpos. Aún hay que determinar si ese aumento de la separación se corresponde con una mayor protección. No obstante, en este contexto pandémico puede parecer poco oportuno esperar tres meses para obtener la inmunidad idónea.
La vacuna se indica a partir de los 18 años -dos más que la indicación de la de Pfizer y el mismo umbral mínimo establecido para la de Moderna- y su eficacia en mayores de 65 años no está tan medida como en otros estudios, si bien casi un 6% de los participantes de los ensayos se encuentran en esta franja de edad, la baja incidencia de infecciones impidió tener datos robustos de eficacia. No obstante, en este grupo poblacional se constató, a través de la medición de anticuerpos, una respuesta adecuada. El perfil de seguridad fue similar con respecto al grupo de 18 a 64 años.
Con todo, la ficha técnica de la vacuna dice que “los datos de eficacia y seguridad son limitados en individuos mayores de 65 años”. Para Pérez Martín parece plausible con esta información que el objetivo de esta vacuna sea una población más joven, si bien “es una estrategia que deben definir los expertos en salud pública británicos. No olvidemos que hay que vacunar a mucha a gente».
Además de los ensayos con los que ha logrado la aprobación británica, la compañía tiene en marcha también un ensayo de combinación de su vacuna con la rusa, conocida como Sputnik V (Ad26), basada también en un vector viral, aunque en este caso humano, con el objetivo de averiguar si mejora así la eficacia de la de AstraZeneca.
Una ventaja diferenciadora de esta vacuna frente, en concreto frente a la de Pfizer+BioNTech, son las condiciones de refrigeración necesarias para su manejo. Así, esta última requiere una temperatura de conservación de unos -75°C±15°C, aunque la multinacional estadounidense ha desarrollado un sistema para facilitar en gran medida la logística de distribución y almacenamiento hasta el punto final de administración de su vacuna gracias a una sofisticada infraestructura, con torre de control incluida, para el seguimiento permanente de los viales. La vacuna aprobada hoy de AstraZeneca -y la de Moderna-, por su parte, pueden mantenerse a una temperatura entre 2º y 8º, con lo que pueden ser almacenadas durante al menos seis meses, transportadas y manipuladas en condiciones de refregeración habituales en la vacunación y, por tanto, administradas con facilidad en los centros sanitarios.
Junto a esta característica favorable, y para lograr la mayor distribución posible de la vacuna, AstraZeneca «ha aprovechado su propia capacidad industrial y se ha asociado con más de 20 colaboradores para la distribución de la vacuna en más de 15 países apoyados por más de 20 centros de analítica para establecer cadenas de suministro en paralelo en un tiempo récord y garantizar así el acceso global a la vacuna, con el objetivo de alcanzar los 3.000 millones de dosis, según explica Per Alfredsson, vicepresidente de Operaciones Biológicas de la compañía.
La vacuna de Janssen una dosis única
‘The New England’ publica resultados de un ensayo en fase 2 con la vacuna Ad26.COV2.S, una de las vacunas que podría administrarse en una sola dosis.
La vacuna de Janssen se basa en un vector viral (adenovirus humano) sin capacidad de replicación.
Sonia Moreno
Jue, 14/01/2021 – 08:00
El análisis intermedio del ensayo en fase 1-2a de la candidata vacunal Ad26SARS-Cov-2 que desarrolla Janssen, filial europea de Johnson & Johnson, revela un buen perfil de seguridad y reactogenidad, así como de inmunogenicidad en un esquema de una dosis y de dos dosis, según acaba de publicar The New England Journal of Medicine.
Meses antes de que Araceli Rosario Hidalgo se convirtiera en la primera española en recibir una vacuna autorizada contra la covid, en España la candidata vacunal de Janssen era la primera inmunización que se administraba a los voluntarios en el contexto de un ensayo, que posteriormente se ha continuado a la fase 3 en varios hospitales de todo el país.
Esta inmunización se basa en un vector viral sin capacidad de replicación –adenovirus humano a diferencia del de chimpancé de la vacuna de Oxford y AstraZeneca- y utiliza la misma plataforma y tecnología que la vacuna de Janssen aprobada para la prevención del Ébola.
Los resultados que recoge ahora The New England se corresponden a un ensayo realizado en doce centros de Bélgica y Estados Unidos, sobre 805 participantes repartidos en un grupo de entre 18 y 55 años, y otro grupo específico de 65 años o más. En el estudio se han recabado datos frente a placebo de la administración de una dosis baja o una dosis alta en un esquema de una sola dosis o de dos dosis.
Como es de esperar en esta fase de ensayo, los criterios de valoración principales fueron la seguridad y la reactogenicidad de cada programa de dosis.
Tras la administración de la primera dosis de vacuna, los eventos adversos más frecuentes fueron fatiga, dolor de cabeza, mialgia y dolor en el lugar de la inyección. El evento adverso sistémico más frecuente fue la fiebre. Los efectos adversos sistémicos fueron menos frecuentes entre los mayores que en los individuos más jóvenes, y en los que recibieron la dosis baja de la vacuna que en los que recibieron la dosis alta. Al contrario de lo que se ha observado con las vacunas de ARN mensajero, la reactogenicidad fue menor después de la segunda dosis.
El trabajo, que ha dirigido la viróloga holandesa Hanneke Schuitemaker, de Janssen, está ahora recopilando datos a largo plazo para comparar el régimen de dosis única con dos dosis, por lo que aún no pueden extraerse conclusiones fehacientes al cotejar ambos regímenes. No obstante, en esta publicación se informa de que una dosis única de Ad26.COV2.S provocó una respuesta humoral fuerte en la mayoría de los que la recibieron, con la presencia de unión a la proteína coronavírica S y anticuerpos neutralizantes en más del 90% de los participantes, independientemente del grupo de edad o de la dosis.
Los autores escriben que “durante 71 días de seguimiento tras la primera dosis, los títulos de anticuerpos aumentaron y se estabilizaron aún más, lo que sugiere la durabilidad de la respuesta inmune provocada por Ad26.COV2.S”.
“Una vacuna eficaz de dosis única para la covid-19 tiene obvias ventajas logísticas sobre una vacuna de dos dosis, especialmente durante una pandemia”, escriben los autores. “Observamos que entre los participantes de 18 y 55 años, una segunda dosis de vacuna el día 57 aumentó aún más el título de anticuerpos, un hallazgo que también estaba en línea con nuestras observaciones recientes en primates no humanos”.
El estudio está analizando ahora si una segunda dosis proporciona un beneficio adicional para mejorar la eficacia o la durabilidad, especialmente en personas de edad avanzada en quienes la respuesta inmune después de la primera dosis tendió a ser moderadamente más baja que la de los participantes más jóvenes.
Para los investigadores los hallazgos presentados en este estudio, junto con los resultados de los estudios preclínicos, respaldan la decisión de avanzar en dos ensayos de fase 3 para evaluar la eficacia de un régimen de dosis única o de dos dosis de la dosis más baja de la vacuna.
La compañía confía en poder anunciar los primeros datos de la fase 3 de su vacuna candidata administrada en dosis única a fines de enero de 2021. Si se demuestra que es eficaz con un buen perfil de seguridad, Janssen espera presentar una solicitud de autorización de uso de emergencia ante la FDA estadounidense poco después.
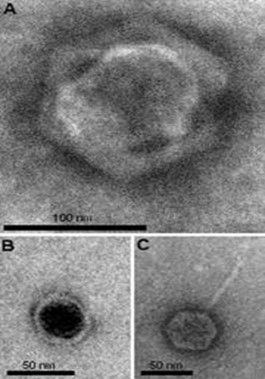
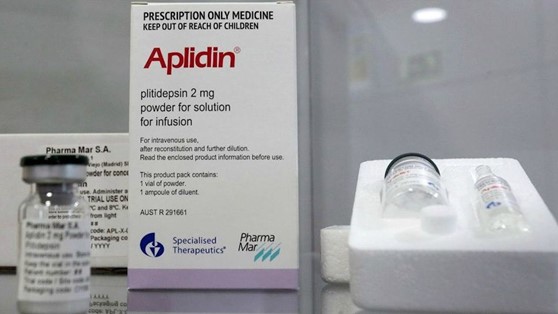
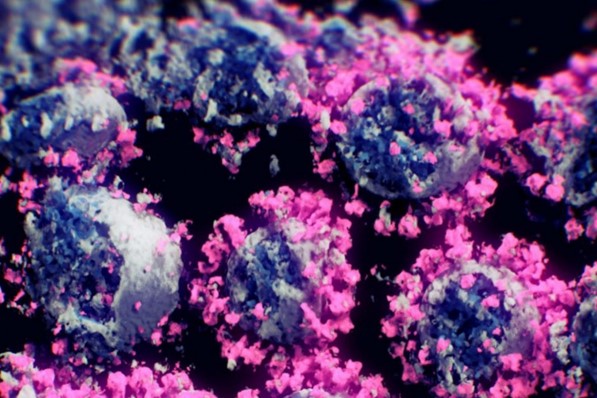
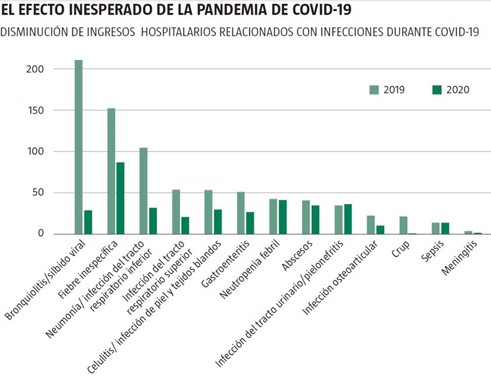
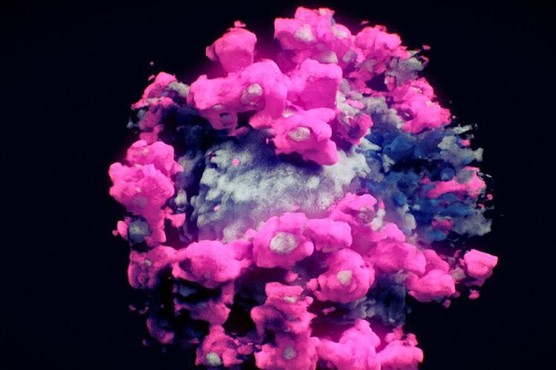

 Mantener la distancia es importante, pero hay que tener en cuenta otros factores de protección para salvarse del contagio.
Mantener la distancia es importante, pero hay que tener en cuenta otros factores de protección para salvarse del contagio.


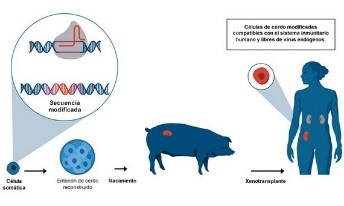
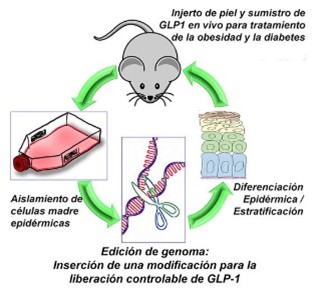

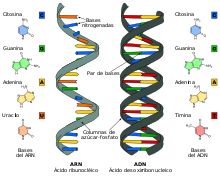
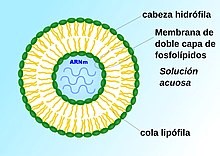
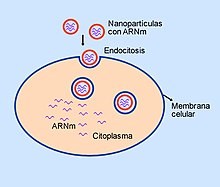
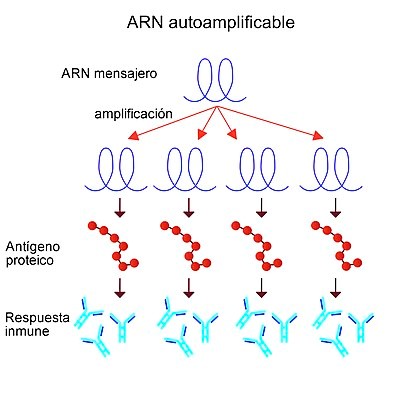
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}