BETA-AMILOIDE
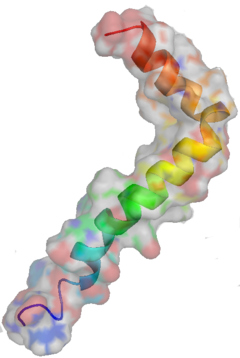
Una de las proteínas mas comprometida en enfermedades plurales. Y sobre todo en la enfermedad de Alzheimer que en nuestros días esta mutinando a multiples enfermos.
A nivel personal y dada mi afición a la actividad infecciosa de una microbiota rota y de los agentes polucionanates “ agentes toxicos e infecciosos”, me invade la idea de Robert Moir 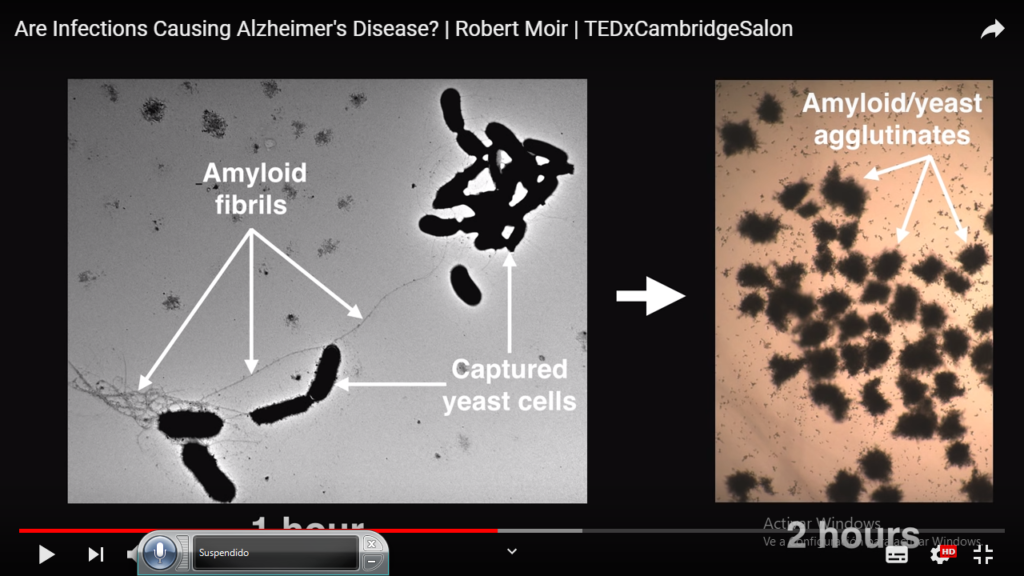 Robert Moir
Robert Moir
La relación entre las enfermedades degenerativas y patógenos, nos podría aclarar, el cambio que ha tenido la patología después de la revolución industrial , y que ha marcado un hito para comprender la etiopatogenia de estas enfermedades. Los trabajos de Kevin Tracey sobre Las enfermedades degenerativas tales como : enfermedad de Parkinson, ELA, esclerosis múltiple, enfermedad de Hungtintom entre otras, comparten, un alteración de la microbiota, una emigración de germenes desde las grandes cavidades orgánicas. Estos germenes se asientan en toda nuestra biología, por supuesto incluyendo el sistema nervioso, una reacción por parte del receptor y un intento de reparación del sistema inmunitario y como consecuencias, el depósito de Macrófagos que inutilizan parcial o totalmente al receptor.
El β-amiloide es un péptido de 36 a 43 aminoácidos que se sintetiza a partir de la proteína precursora amiloidea (APP). Aunque generalmente se alude a su relación con la enfermedad de Alzheimer, se ha encontrado evidencia de que tiene múltiples actividades no asociadas con esta enfermedad.1
En condiciones no asociadas con Alzheimer incluyen: la activación de quinasas,23 la protección contra estrés oxidativo,45 la regulación del transporte de colesterol,67 (actuando como un factor de transcripción)89 y la actividad antimicrobiana, vinculada con su acción proinflamatoria.10
Este péptido es el principal componente de las placas seniles (depósitos que se encuentran en el cerebro de los pacientes con la enfermedad de Alzheimer), aunque existen placas similares en otras demencias: en la Demencia de cuerpos de Lewy y en la miositis por cuerpos de inclusión (enfermedad muscular). El β-amiloide también puede formar agregados que cubren vasos sanguíneos cerebrales en la angiopatía amiloide cerebral. Las placas se forman por una red irregular de agregados fibrilares llamadas fibras amiloides,11 un plegamiento proteico asociado a otros péptidos como los priones, proteínas patógenas alteradas, que tienen un plegamiento incorrecto. Investigaciones recientes sugieren que las formas solubles de las estructuras oligoméricas del péptido pueden ser agentes causales del desarrollo de la enfermedad de Alzheimer.12
El β-amiloide se forma por la división secuencial del precursor proteico amiloide (APP), una glicoproteína transmembrana con una función indeterminada. El APP puede ser procesado a partir de las α-secretasa, β-secretasa y γ-secretasa. En primera instancia el β-amiloide se forma por la acción sucesiva de las secretasas β y γ. Ambas producen el extremo C-terminal del péptido y a su vez se incorporan a la región transmembrana del APP con el objetivo de generar isoformas de 36 a 43 aminoácidos. Las isoformas más comunes son Aβ40 y Aβ42, donde la isoforma más corta se genera debido a la escisión que ocurre en el retículo endoplasmático, mientras que la isoforma larga se escinde en el área trans-Golgi.13 La isoforma Aβ40 es la más habitual. La Aβ42 es más fibrogénica y, por lo tanto, está asociada con el desarrollo de ciertas enfermedades. Mutaciones en el APP asociados con estadios iniciales de Alzheimer se relacionan con un aumento en la producción de Aβ42 por lo que la terapia para combatir el Alzheimer se basa en regular la actividad de las secretasas β y γ para que la producción de A40 sea mayor.14
Las placas seniles descritas por Alois Alzheimer son el resultado de la acumulación y la precipitación anormal de péptido βA fuera de la célula. Debido a su mayor agregación e insolubilidad el Aβ42 es más abundante en las placas que el Aβ40. La clasificación más utilizada es la morfológica, que distingue entre dos tipos de placas: las difusas y las densas, según su capacidad de tinción con Rojo Congo y Tioflavina-S.
Esta división es relevante en la enfermedad de Alzheimer, ya que las placas densas tioflavina-S positivas están asociadas con: efectos deletéreos en el neuropilo, incremento de la curvatura y distrofia de las neuritas (axones y dendritas), pérdida de sinapsis y de neuronas, y activación y reclutamiento de astrocitos y microglia.
Las placas difusas están presentes en el cerebro de personas mayores cuyas capacidades cognitivas están intactas, por el contrario, las placas densas (particularmente aquellas con distrofia en las neuritas) solo se encuentran en pacientes con enfermedad de Alzheimer. A pesar de esto, los límites entre el envejecimiento normal y la demencia de Alzheimer no están claros, pues hay personas con capacidades cognitivas conservadas en las que se pueden encontrar depósitos de amiloide.
Mutaciones autosómicas dominantes en el APP causan, mediante un mecanismo hereditario, la enfermedad de Alzheimer (EA), en un 10% de los casos. La mayoría de los casos de EA no son causadas por este tipo de mutaciones y no siguen un patrón hereditario.16 Un aumento en los niveles totales de Aβ o un aumento en la concentración tanto de Aβ40 y Aβ4217 se relaciona completamente con la patogenia de ambos tipos de EA esporádica. Debido a que la isoforma Aβ42 tiene una naturaleza más hidrofóbica se le considera la forma más amiloidogénica del péptido. Sin embargo, la secuencia central KLVFFAAE es la responsable de formar amiloide por su cuenta y probablemente forma la cubierta de fibrilla. La “hipótesis del amiloide” propone que las placas son las responsables de causar la EA sin embargo esta teoría, a pesar de tener una gran aceptación en el sector científico, no está completamente fundamentada. Los depósitos intracelulares de la proteína tau también se observan como signo en el desarrollo de la EA. Los oligómeros que forma la vía del amiloide podrían ser las especies citotóxicas.18Una hipótesis alternativa es que los oligómeros de amiloide son responsables del desarrollo de la enfermedad. Los roedores que han sido modificados genéticamente expresan oligómeros, pero no expresan placas (APPE693Q) que desencadenan la enfermedad.19
AMILOIDOSIS FAMILIAR HEREDITARIA
La amiloidosis familiar hereditaria fue reconocida por la caracterización bioquímica de la proteína amiloide depositada. Los primeros investigadores de la amiloidosis familiar clasificaban esta enfermedad en función del órgano predominantemente afectado, como amiloidosis neuropática1 o amiloidosis renal2. Costa, al identificar en 1978 una variante de la prealbúmina, actualmente llamada transtirretina, como la proteína presente en la sustancia amiloide de la polineuropatía amiloidótica familiar (PAF) de tipo portugués descrita por Andrade, abrió el camino de la caracterización bioquímica de las diversas formas de amiloidosis hereditarias3. En la última década, se han descrito otras formas de amiloidosis familiar y se han identificado sus proteínas anómalas precursoras y las mutaciones asociadas: amiloidosis renal, hepática y polineuropática en los EE.UU. y Reino Unido, debida a una variante anómala de la apoproteína AI4; amiloidosis corneal y encefálica finlandesa debida a una gelsolina anómala5; amiloidosis renal y hepática por una anomalía de la cadena alfa del fibrinógeno en México y los EE.UU.6; amiloidosis renal británica por una lisozima anómala7; y amiloidosis cerebrovascular con hemorragia cerebral en Islandia y Holanda debida al depósito de una cistatina C atípica8. Además, la descripción de la amiloidosis de tipo AA como complicación de la fiebre mediterránea familiar (FMF), y la identificación de la mutación genética característica de esta enfermedad, descrita por un consorcio internacional de investigadores y publicada en 19979,10 ha completado la nueva clasificación de las amiloidosis familiares hereditarias, basada en la proteína anómala que origina el depósito de amiloide Las amiloidosis familiares son enfermedades de transmisión genética autosómica dominante, excepto la amiloidosis asociada a la FMF, cuya transmisión es recesiva. Debido a una mutación en el gen que codifica la síntesis de una proteína, ésta sufre una anomalía estructural que le confiere una alteración de su solubilidad, y tras pasar al torrente circulatorio se deposita en ciertos órganos y tejidos. Aunque este depósito es progresivo a lo largo de toda la vida, en muchas formas de amiloidosis hereditaria familiar las manifestaciones clínicas de la enfermedad no comienzan hasta la tercera o cuarta década de la vida, o incluso más tardíamente. Su presentación clínica es muy variada, lo que puede dar lugar a problemas diagnósticos. Debido a su heterogeneidad genética y bioquímica, mutaciones diferentes son capaces de producir una misma forma clínica de enfermedad11 y por el contrario una mutación idéntica puede dar lugar a manifestaciones clínicas variables de un individuo a otro12. Las dos formas de amiloidosis familiares más prevalentes en nuestro entorno son la PAF y la FMF, que se asocia en ocasiones a amiloidosis renal.
FIEBRE MEDITERRANEA FAMILIAR
La FMF es una enfermedad hereditaria autosómica recesiva que se caracteriza por crisis recurrentes y breves de fiebre, dolor e inflamación de una o varias serosas. La fiebre y el dolor abdominal son los síntomas más frecuentes durante las crisis, aunque también puede haber dolores articulares y torácicos, así como manifestaciones cutáneas. En los períodos intercrisis los pacientes están asintomáticos. La amiloidosis es la complicación más importante de la FMF, y suele ser causa de muerte en los casos en que se presenta.
En 1992 se localizó la alteración genética asociada a la FMF en el brazo corto del cromosoma 1613. En 1997 dos grupos independientes consiguieron clonar y caracterizar el gen de la FMF, denominándolo MEFV9,10. Este gen codifica una proteína que fue denominada «marenostrina» o «pirina» según el equipo investigador. Hasta ahora se han descrito varias mutaciones diferentes en el exón 10 del gen MEFV, siendo las más frecuentes las denominadas M6801, M694V, V726A14.
La patogenia de esta enfermedad sigue siendo confusa y son varias las teorías que se han propuesto para explicar la diversa sintomatología que la acompaña. La marenostrina/pirina, que se expresa casi exclusivamente en los leucocitos neutrófilos, podría causar una activación descontrolada de estas células y su migración a los tejidos serosos15.
Los pacientes con FMF son descendientes de judíos sefarditas, askenazis, armenios, árabes y turcos. En nuestro país se ha identificado en Mallorca un grupo de enfermos entre los descendientes de judíos conversos. En un 30% de ellos se ha identificado un haplotipo genético similar a otros pacientes de otras zonas geográficas16. De manera aislada se han diagnosticado pacientes en Gerona, Toledo, Madrid y ciertas áreas de Andalucía.
La amiloidosis asociada a la FMF se debe al depósito de la proteína amiloide de tipo AA, que es un producto de la degradación de la proteína sérica precursora del amiloide (SAP), un reactante de fase aguda sintetizado por el hígado. El depósito más frecuente e intenso de amiloide se encuentra en los riñones, glándulas adrenales, intestino, bazo e hígado; más raramente se deposita en el pulmón, el tiroides, el corazón y los testículos. La proteína amiloide, de configuración fibrilar, infiltra la capa íntima y la media de las arteriolas, así como el parénquima renal. En el hígado los depósitos de amiloide están limitados a las paredes de la vena porta. En la mayoría de los casos, la amiloidosis se desarrolla antes de los 40 años de edad. La manifestación clínica más frecuente de la amiloidosis asociada a la FMF es la aparición de un síndrome nefrótico, con insuficiencia renal posterior. El síndrome de malabsorción intestinal y la insuficiencia adrenal son poco frecuentes, y a diferencia de otras formas de amiloidosis no se produce una afectación neuropática14. El diagnóstico de la amiloidosis se puede realizar mediante biopsia rectal, renal o de grasa abdominal. Se ha observado que la prevalencia de la amiloidosis asociada a la FMF es diferente en los diversos grupos étnicos, siendo más elevada en judíos del norte de África y de Turquía y más baja en judíos iraquíes, askenazis y árabes. La administración prolongada de colchicina ha disminuido significativamente la incidencia de amiloidosis en los pacientes con FMF17. El pronóstico de la FMF depende de la aparición de amiloidosis. En función de ello, se describen dos fenotipos de la FMF. En el fenotipo I, el más frecuente, las manifestaciones clínicas preceden a la amiloidosis, y por lo general se observa una disminución en el número y la intensidad de las crisis conforme avanza la edad del paciente. En el fenotipo II, la amiloidosis es la primera o única manifestación de la enfermedad. Se ha hallado una correlación entre el genotipo y el fenotipo. En los homocigotos M694V, la FMF se caracteriza por un inicio más temprano, con crisis más frecuentes, un mayor número de articulaciones afectadas, y un riesgo mayor de aparición de amiloidosis. Serán necesarios más estudios para confirmar la correlación entre el genotipo y el fenotipo18.
Hasta hace poco tiempo, el diagnóstico de la FMF se basaba en las manifestaciones clínicas, el origen étnico, la historia familiar y la respuesta a la colchicina. El test de provocación con metaraminol no estaba exento de efectos secundarios19. Actualmente, la clonación del gen MEFV permite disponer de una prueba diagnóstica nueva y fiable para esta enfermedad. Las tres principales mutaciones hasta ahora descritas M6801, M694V, V726A están presentes en la mayoría de los pacientes con FMF. En los casos en que no se consigue identificar la mutación, el diagnóstico se sigue basando en la clínica. Recientemente se ha publicado una nueva propuesta de criterios diagnósticos clínicos, en un intento de mejorar la precisión diagnóstica20. En las primeras crisis agudas de la enfermedad, y en ausencia de antecedentes familiares, es difícil hacer el diagnóstico de la FMF, y a menudo se confunde con un cuadro de abdomen agudo, lo que con frecuencia supone la indicación de laparotomía.
La administración prolongada de colchicina, a dosis de 0,5 mg/30 kg/día, consigue disminuir la frecuencia y la intensidad de las crisis y prevenir la aparición de amiloidosis21,17. No existe ningún fármaco eficaz para el tratamiento sintomático durante las crisis. El trasplante renal debe realizarse cuando existe insuficiencia renal avanzada por amiloidosis. La administración de colchicina en los pacientes trasplantados evita la reaparición de amiloidosis en el injerto17.
POLINEUROPATIA AMILOIDOTICA FAMILIAR
La PAF se debe al depósito extracelular de una variante anómala de la transtirretina (TTR), una proteína plasmática también llamada prealbúmina sintetizada predominantemente en el hígado (aunque también se sintetiza en el plexo coroideo y en la retina), cuya función fisiológica es el transporte de hormonas tiroideas y del complejo formado por la vitamina A (retinol) con su proteína transportadora específica22-24. Aunque la PAF fue descrita inicialmente en varias familias portuguesas, y su foco endémico más importante está en Portugal, también se han descrito variantes de la misma enfermedad en Suecia, Finlandia, Japón, EE.UU., Brasil, y en ciertas áreas del Mediterráneo como Italia, Grecia, Turquía e Israel. En España también hay un foco endémico, localizado en Mallorca25. El defecto genético asociado a esta enfermedad se transmite según un patrón autosómico dominante, y consiste en una mutación del gen que codifica la síntesis de la TTR, localizado en el cromosoma 18. La mutación más frecuente es la que provoca la PAF clásica descrita por Andrade en Portugal (PAF tipo I), y consiste en la sustitución simple de una guanina por una adenina en el gen de la TTR26, lo que provoca la síntesis de una TTR anómala en la que una valina es sustituida por una metionina en la posición 30 de la cadena peptídica (TTR Met30)27. Este cambio estructural confiere a la TTR una especial tendencia a depositarse en forma de amiloide28.
En los últimos años, se han descrito más de 50 mutaciones de la TTR29. Se ha observado que la mayoría de las diferentes variantes anómalas de TTR provocan una forma clínica diferente de PAF, con un curso clínico y una velocidad de progresión específicas, algunas más rápidas y graves y otras más lentas y leves. También se han descrito mutaciones de la TTR no amiloidogénicas, una de las cuales, la TTR Met119, muestra una mayor estabilidad que la TTR normal30, e incluso tiene un papel relativamente «protector», ya que la presencia simultánea de las mutaciones Met30 y Met119 en un mismo paciente se asocia a una enfermedad de progresión más lenta31.
La PAF tipo I se manifiesta como una polineuropatía mixta y progresiva que suele iniciarse en la tercera o cuarta décadas de la vida, aunque hay casos más precoces y también más tardíos. La polineuropatía, inicialmente sensitiva y posteriormente también motora y autonómica, se inicia en extremidades inferiores distales, tiene una evolución ascendente, y más tarde afecta a extremidades superiores, tronco y nervios craneales1,32. En los últimos años se ha caracterizado con más precisión la afectación sistémica de la enfermedad, especialmente la digestiva, cardiocirculatoria, renal y ocular. La alteración digestiva fundamental es la dismotilidad, que inicialmente causa estreñimiento y posteriormente diarrea crónica y malabsorción por sobrecrecimiento bacteriano. En el miocardio el depósito de amiloide altera relativamente poco la capacidad contráctil, pero da una imagen ecocardiográfica brillante muy llamativa y característica33. En los riñones, el depósito de amiloide puede provocar proteinuria e insuficiencia renal34. En los ojos, además de opacidades en el cuerpo vítreo con relativamente poca repercusión en la agudeza visual35, pueden aparecer hendiduras pupilares, queratoconjuntivitis seca, anomalías de los vasos conjuntivales y glaucoma36.
La enfermedad tiene un curso inexorablemente progresivo, y causa la muerte 7-15 años después del inicio de las manifestaciones clínicas37. La causa de muerte más frecuente es la aparición de complicaciones sépticas en el contexto de un paciente debilitado, malnutrido, hipotenso y cardiópata. En los últimos años se han identificado diversos factores predictivos de la velocidad de progresión y del pronóstico de la enfermedad. Los casos que comienzan clínicamente a una edad más temprana suelen evolucionar más rápidamente. Éste suele ser el caso de los pacientes varones cuya madre se encuentra afectada de la enfermedad (o es portadora sana de la TTR anómala, sin amiloide tisular y sin síntomas)38. Son signos de mal pronóstico vital la edad avanzada, la presencia de cardiomiopatía37 y, muy especialmente, la aparición de malabsorción y malnutrición39.
El único tratamiento eficaz para la PAF es el trasplante hepático (TH). El primer TH en un paciente con PAF, realizado en Suecia en 1990, permitió comprobar que, tras el trasplante, la TTR anómala pasa a ser prácticamente indetectable en sangre y que se detiene la progresión de la enfermedad40, observándose una lenta mejoría sintomática e incluso una progresiva disminución del depósito de amiloide41. En 1993, un simposio internacional, celebrado en Suecia, declaró que el TH es el único tratamiento válido para los pacientes con PAF, y estableció un registro mundial de TH en pacientes con PAF, con base en Huddinge (Suecia). Desde 1990 se han realizado TH en pacientes con PAF en numerosos hospitales en Europa, América, Australia y Japón. En mayo de 1998, el registro mundial contabilizaba casi 300 TH realizados por esta indicación en todo el mundo, de los cuales 35 habían sido realizados en España, la mayoría de ellos en pacientes de origen mallorquín. Los países en los que se habían realizado más trasplantes en pacientes con PAF eran, por este orden, Portugal, Suecia, Francia, España y Estados Unidos. La supervivencia postrasplante de estos pacientes ha demostrado ser similar a la obtenida en otros grupos de pacientes trasplantados por hepatopatías crónicas, alcanzando más de un 70% al año y más de un 60% a los 5 años del TH42.
Recientemente se ha publicado en esta revista una excelente revisión de las indicaciones y resultados del TH en pacientes con PAF43. La experiencia acumulada en estos años ha resuelto muchas de las incógnitas que surgieron cuando se realizaron los primeros TH por PAF44, y han confirmado que el TH no sólo detiene la progresión de la PAF, sino que permite a los pacientes iniciar una lenta y progresiva mejoría de los síntomas digestivos y neuropáticos, aunque es difícil objetivar esta mejoría en pruebas electrofisiológicas45-47. Recientemente se ha descrito la disminución del depósito perineural de amiloide y la regeneración de las fibras de mielina después del TH48. Este hallazgo confirma que la sustancia amiloide depositada no es totalmente insoluble, sino que puede ser catabolizada y eliminada in vivo (y que su acumulación es un proceso dinámico en el que la velocidad de depósito es mayor que la capacidad de eliminación tisular del amiloide). Sin embargo, en algunos pacientes se ha observado que, tras el TH, la cardiopatía puede continuar progresando y evolucionar a insuficiencia cardíaca y muerte súbita49. Este fenómeno sólo se ha observado en pacientes con formas de PAF diferentes de la PAF tipo I (con variantes de la TTR distintas de la TTR Met30).
Actualmente se considera que el TH debe indicarse en una fase precoz de la enfermedad, cuando los síntomas de la PAF comienzan a limitar la actividad diaria o la calidad de vida del paciente. Hay que evitar hacer el TH en una fase demasiado evolucionada de la enfermedad, porque se asocia una mayor morbimortalidad postrasplante. Además, en los supervivientes la calidad de vida postrasplante seguiría siendo pobre. Por otra parte, los pacientes con PAF tienen un mayor riesgo de hipotensión grave durante el acto quirúrgico del trasplante, porque tienen alterados el reflejo vasomotor y la respuesta a los fármacos vasopresores50. Recientemente se han identificado varios factores asociados a una alta mortalidad postrasplante de los pacientes con PAF: el tiempo de evolución de la enfermedad, la presencia de malabsorción y malnutrición, y la insuficiencia renal pretrasplante39,51.
Una de las novedades más llamativas en los últimos años ha sido la realización del denominado domino liver transplantation (o TH secuencial), que consiste en el aprovechamiento del hígado nativo del paciente con PAF sometido a TH, para trasplantarlo a su vez a otro paciente con hepatopatía crónica. Los hígados de los pacientes con PAF son metabólicamente normales, salvo por la producción de la TTR anómala, y el tiempo de incubación hasta la aparición de amiloidosis (2-3 décadas) puede ser superior a la supervivencia esperable de los hepatópatas receptores de TH con una edad relativamente avanzada (60 años o más). El TH domino contribuye a no empeorar las largas listas de espera para TH, y ha sido ya aceptado en algunos centros de TH, que ya están ofreciendo los hígados de los pacientes con PAF a otros pacientes en lista de espera, previa información de los riesgos potenciales. El registro mundial de TH en pacientes con PAF reporta, en su informe de 1997, 10 casos de trasplante domino realizados (ocho en Portugal y dos en Suecia)42,52, pero esta práctica ya se ha extendido a otros países (Reino Unido o Canadá) sin que se hayan reportado, hasta el momento, efectos nocivos a corto plazo en los receptores de hígados procedentes de pacientes con PAF53,54.
Dado que el TH es un procedimiento muy agresivo, y que no todos los pacientes con PAF pueden beneficiarse de un TH, se han seguido investigando otros posibles tratamientos para la PAF. Recientemente se ha ensayado un procedimiento para extraer de la sangre la TTR anómala y evitar su depósito, mediante hemofiltración a través de microcapilares en cuyas paredes hay anticuerpos específicos que captan la TTRMet30. Aunque esta inmunohemofiltración disminuye transitoriamente la concentración de TTR anómala en sangre, no ha demostrado un efecto beneficioso a corto plazo, sino que por el contrario ha provocado efectos indeseables como la disminución de las concentraciones séricas de hormonas tiroideas y síntomas de hipovitaminosis A, por lo que este tratamiento experimental, inicialmente prometedor, no ha sido aceptado aún para su aplicación generalizada en clínica55,56.
Otra nueva estrategia terapéutica en fase experimental, potencialmente aplicable a otras formas de amiloidosis, es aumentar el catabolismo de la sustancia amiloide ya depositada. Recientemente se ha observado que la administración de 4′-yodo-4′-deoxidoxorrubicina (I-DOX), un derivado del citostático doxorrubicina con una especial afinidad por la sustancia amiloide, acelera la disolución del amiloide tisular en los pacientes con amiloidosis primaria o asociada a mieloma múltiple (amiloidosis de tipo AL), y en algunos casos ha conseguido estabilizar la enfermedad o incluso mejorar los síntomas57. El I-DOX tiene unos efectos tóxicos que limitan su posible utilización. No obstante, podría ser un punto de partida para el diseño de otros fármacos disolutivos del amiloide más tolerables y efectivos. Otra estrategia terapéutica podría basarse en el estudio de la estructura y las características fisicoquímicas de la proteína sérica precursora de amiloide (serum amyloid protein, SAP), que es común a todas las formas de amiloidosis conocidas. Si se hallase un fármaco que impidiese la adhesión de la SAP a las proteínas precursoras específicas de cada forma de amiloidosis, quizá podría conseguirse una profilaxis eficaz de todas las amiloidosis sistémicas. Se ha logrado crear un modelo experimental de amiloidosis en animales de laboratorio58, lo que facilitará la realización de estudios básicos sobre la amiloidogénesis y el ensayo de nuevas estrategias terapéuticas experimentales59.
Proteína amiloide hepática puede ser causante de la enfermedad de Alzheimer La proteína amiloide fabricada en el hígado puede causar neurodegeneración, según un nuevo estudio realizado por investigadores de la Universidad de Curtin en Bentley (Australia), y publicado en PLOS Biology.
Los investigadores confirman que los depósitos de beta-amiloide (A-beta) en el cerebro son una de las características patológicas de la enfermedad de Alzheimer (EA) y están implicados en la neurodegeneración, tanto en pacientes humanos como en modelos animales. Pero la A-beta también está presente en los órganos periféricos, y los niveles de A-beta en sangre se correlacionan con la carga de amiloide cerebral y el deterioro cognitivo, lo que plantea la posibilidad de que la a-beta producida periféricamente pueda contribuir a la enfermedad. Probar esta hipótesis ha sido difícil, ya que el cerebro también produce A-beta, y distinguir la proteína de las dos fuentes es un reto.
En modelos murinos, los autores demostraron que la proteína era transportada en la sangre por lipoproteínas ricas en triglicéridos, al igual que en los humanos, y pasaba de la periferia al cerebro. Comprobaron que desarrollaban neurodegeneración y atrofia cerebral, lo que iba acompañado de inflamación neurovascular y disfunción de los capilares cerebrales, ambas cosas comúnmente observadas en la enfermedad de Alzheimer.
Los autores concluyen que la A-beta producida en el hígado es un potencial contribuyente a la enfermedad humana. Si esa contribución es significativa, los hallazgos podrían tener importantes implicaciones para la comprensión de la enfermedad de Alzheimer.
BIBLIOGRAFÍA
Lahiri DK, Ma↑loney B (septiembre de 2010). «Beyond the signaling effect role of amyloid–β42 on the processing of AβPP, and its clinical implications». Exp. Neurol. 225 (1): 51-4. PMC 2922469. PMID 20451519. doi:10.1016/j.expneurol.2010.04.018.
Bogoyevitch MA, Boehm I, Oakley A, Ketterman AJ, Barr RK (marzo de 2004). «Targeting the JNK MAPK cascade for inhibition: basic science and therapeutic potential». Biochim. Biophys. Acta 1697 (1–2): 89-101. PMID 15023353. doi:10.1016/j.bbapap.2003.11.016.
Tabaton M, Zhu X, Perry G, Smith MA, Giliberto L (enero de 2010). «Signaling Effect of Amyloid-β42 on the Processing of AβPP». Exp. Neurol. 221 (1): 18-25. PMC 2812589. PMID 19747481. doi:10.1016/j.expneurol.2009.09.002.
Zou K, Gong JS, Yanagisawa K, Michikawa M (junio de 2002). «A novel function of monomeric amyloid beta-protein serving as an antioxidant molecule against metal-induced oxidative damage». J. Neurosci. 22 (12): 4833-41. PMID 12077180.
Baruch-Suchodolsky R, Fischer B (mayo de 2009). «Abeta40, either soluble or aggregated, is a remarkably potent antioxidant in cell-free oxidative systems». Biochemistry 48 (20): 4354-70. PMID 19320465. doi:10.1021/bi802361k.
Yao ZX, Papadopoulos V (octubre de 2002). «Function of beta-amyloid in cholesterol transport: a lead to neurotoxicity». FASEB J. 16 (12): 1677-9. PMID 12206998. doi:10.1096/fj.02-0285fje.
Igbavboa U, Sun GY, Weisman GA, He Y, Wood WG (agosto de 2009). «Amyloid β-Protein Stimulates Trafficking of Cholesterol and Caveolin-1 from the Plasma Membrane to the Golgi Complex in Mouse Primary Astrocytes». Neuroscience 162 (2): 328-38. PMC 3083247. PMID 19401218. doi:10.1016/j.neuroscience.2009.04.049.
Maloney B, Lahiri DK (junio de 2011). «The Alzheimer’s amyloid β-peptide (Aβ) binds a specific DNA Aβ-interacting domain (AβID) in the APP, BACE1, and APOE promoters in a sequence-specific manner: Characterizing a new regulatory motif». Gene 488 (1–2): 1-12. PMID 21699964. doi:10.1016/j.gene.2011.06.004.
Bailey JA, Maloney B, Ge YW, Lahiri DK (junio de 2011). «Functional activity of the novel Alzheimer’s amyloid β-peptide interacting domain (AβID) in the APP and BACE1 promoter sequences and implications in activating apoptotic genes and in amyloidogenesis». Gene 488 (1–2): 13-22. PMID 21708232. doi:10.1016/j.gene.2011.06.017.
Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, Burton MA, Goldstein LE, Duong S, Tanzi RE, Moir RD (2010). «The Alzheimer’s Disease-Associated Amyloid β-Protein Is an Antimicrobial Peptide». En Bush, Ashley I., ed. PLoS ONE 5 (3): e9505. PMC 2831066. PMID 20209079. doi:10.1371/journal.pone.0009505.
Parker MH, Reitz AB (2000). «Assembly of β-Amyloid Aggregates at the Molecular Level». Chemtracts-Organic Chemistry 13 (1): 51-56.
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ (agosto de 2008). «Amyloid β-Protein Dimers Isolated Directly from Alzheimer Brains Impair Synaptic Plasticity and Memory». Nat. Med. 14 (8): 837-42. PMC 2772133. PMID 18568035. doi:10.1038/nm1782. Resumen divulgativo – Fox News.
Hartmann T, Bieger SC, Brühl B, Tienari PJ, Ida N, Allsop D, Roberts GW, Masters CL, Dotti CG, Unsicker K, Beyreuther K (septiembre de 1997). «Distinct sites of intracellular production for Alzheimer’s disease A beta40/42 amyloid peptides». Nat. Med. 3 (9): 1016-20. PMID 9288729. doi:10.1038/nm0997-1016.
Yin YI, Bassit B, Zhu L, Yang X, Wang C, Li YM (agosto de 2007). «γ-Secretase Substrate Concentration Modulates the Aβ42/Aβ40 Ratio: Implications for Alzheimer’s disease». J. Biol. Chem. 282 (32): 23639-44. PMID 17556361. doi:10.1074/jbc.M704601200.
Serrano-Pozo A, Forsch M.P, Masliah E, Hyman B.T. «Neuropathological alterations in Alzheimer disease». Cold Spring Harb Perspect Med. 1 (1). doi:10.1101/cshperspect.a006189.
Maslow K (marzo de 2008). «2008 Alzheimer’s disease facts and figures». Alzheimers Dement 4 (2): 110-33. PMID 18631956. doi:10.1016/j.jalz.2008.02.005.
Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J (septiembre de 1999). «Soluble Amyloid β Peptide Concentration as a Predictor of Synaptic Change in Alzheimer’s Disease». Am. J. Pathol. 155 (3): 853-
Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG (abril de 2003). «Common structure of soluble amyloid oligomers implies common mechanism of Pathogenesis». Science 300 (5618): 486- 9. PMID 12702875. doi:10.1126/science.1079469.
Gandy S, Simon AJ, Steele JW, Lublin AL, Lah JJ, Walker LC, Levey AI, Krafft GA, Levy EF, Checler F, Glabe C, Bilker W, Abel T, Schmeidler J, Ehrlich ME (2010). «Days-to-criterion as an indicator of toxicity associated with human Alzheimer amyloid-β oligomers». Annals of Neurology 67 (6): 220-30. PMC 3094694. PMID 20641005. doi:10.1002/ana.22052. Resumen divulgativo – Drug Discovery and Development.
Zhang S, Iwata K, Lachenmann MJ, Peng JW, Li S, Stimson ER, Lu Y, Felix AM, Maggio JE, Lee JP (2000). «The Alzheimer’s Peptide Aβ Adopts a Collapsed Coil Structure in Water». Journal of Structural Biology (en inglés) 130 (2–3): 130-141. PMID 10940221. doi:10.1006/jsbi.2000.4288.
Yang M, Teplow DB (2008). «Amyloid β-Protein Monomer Folding: Free-Energy Surfaces Reveal Alloform-Specific Differences». Journal of Molecular Biology (en inglés) 384 (2): 450-464. PMC 2673916. PMID 18835397. doi:10.1016/j.jmb.2008.09.039.
Sgourakis NG, Yan Y, McCallum SA, Wang C, Garcia AE (2007). «The Alzheimer’s peptides Aβ40 and 42 adopt distinct conformations in water: A combined MD / NMR study». Journal of Molecular Biology (en inglés) 368 (5): 1448-1457. PMC 1978067. PMID 17397862. doi:10.1016/j.jmb.2007.02.093.
Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, Elliott JI, Van Nostrand WE, Smith SO (mayo de 2010). «Structural conversion of neurotoxic amyloid-beta(1-42) oligomers to fibrils». Nat. Struct. Mol. Biol. (en inglés) 17 (5): 561-7. PMC 2922021. PMID 20383142. doi:10.1038/nsmb.1799.
Yu L, Edalji R, Harlan JE, Holzman TF, Lopez AP, Labkovsky B, Hillen H, Barghorn S, Ebert U, Richardson PL, Miesbauer L, Solomon L, Bartley D, Walter K, Johnson RW, Hajduk PJ, Olejniczak ET (2009). «Structural Characterization of a Soluble Amyloid β-Peptide Oligomer». Biochemistry (en inglés) 48 (9): 1870-1877. PMID 19216516. doi:10.1021/bi802046n.
Citron M (septiembre de 2004). «Strategies for disease modification in Alzheimer’s disease». Nat. Rev. Neurosci. 5 (9): 677-85. PMID 15322526. doi:10.1038/nrn1495.
Lashuel HA, Hartley DM, Balakhaneh D, Aggarwal A, Teichberg S, Callaway DJ (noviembre de 2002). «New class of inhibitors of amyloid-beta fibril formation. Implications for the mechanism of pathogenesis in Alzheimer’s disease». J. Biol. Chem. 277 (45): 42881-90. PMID 12167652. doi:10.1074/jbc.M206593200.
Michael H. Parker, Robert Chen, Kelly A. Conway, Daniel H. S. Lee; Chi Luoi, Robert E. Boyd, Samuel O. Nortey, Tina M. Ross, Malcolm K. Scott, Allen B. Reitz (2002). «Synthesis of (+)-5,8-Dihydroxy-3R-methyl-2R-(dipropylamino)-1,2,3,4-tetrahydro-naphthalene: An Inhibitor of β-Amyloid1-42 Aggregation». Bioorg. Med. Chem 10 (11): 3565-3569. PMID 12213471. doi:10.1016/S0968-0896(02)00251-1.
Pappolla M, Bozner P, Soto C, Shao H, Robakis NK, Zagorski M, Frangione B, Ghiso J (marzo de 1998). «Inhibition of Alzheimer’s beta fibrillogenesis by melatonin». J Biol Chem 273 (13): 7185-7188. PMID 9516407. doi:10.1074/jbc.273.13.7185.
Lahiri DK, Chen DM, Lahiri P, Bondy S, Greig NH (noviembre de 2005). «Amyloid, cholinesterase, melatonin, and metals and their roles in aging and neurodegenerative diseases». Ann. N. Y. Acad. Sci. 1056: 430-49. PMID 16387707. doi:10.1196/annals.1352.008.
Wang XC, Zhang YC, Chatterjie N, Grundke-Iqbal I, Iqbal K, Wang JZ (junio de 2008). «Effect of melatonin and melatonylvalpromide on beta-amyloid and neurofilaments in N2a cells». Neurochem. Res. 33 (6): 1138-44. PMID 18231852. doi:10.1007/s11064-007-9563-y.
Saltar a:a b c d Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM (noviembre de 2009). «Amyloid-β Dynamics are Regulated by Orexin and the Sleep-Wake Cycle». Science 326 (5955): 1005-7. PMC 2789838. PMID 19779148. doi:10.1126/science.1180962.
doi 10.1523/JNEUROSCI.4540-04.2005
Rekas A, Jankova L, Thorn DC, Cappai R, Carver JA (14 de noviembre de 2007). «Monitoring the prevention of amyloid fibril formation by α‐crystallin» (PDF). The FEBS Journal 274. doi:10.1111/j.1742-4658.2007.06144.x. Consultado el 7 de abril de 2022.
Gengler S, Gault VA, Harriott P, Hölscher C. Estos procesos de agregación también pueden ser estudiados en las bicapas lipídicas.
Sanghera N, Swann MJ, Ronan G, Pinheiro TJ (octubre de 2009). «Insight into early events in the aggregation of the prion protein on lipid membranes». Biochim. Biophys. Acta 1788 (10): 2245-51. PMID 19703409. doi:10.1016/j.bbamem.2009.08.005.
Lam V, Takechi R, Hackett MJ, Francis R, Bynevelt M, et al.PLoS Biology 19(9): e3001358. https://doi.org/10.1371/journal.pbio.3001358
Universidad de Curtin en Bentley (Australia), y publicado en PLOS Biology.