TERAPIA EXPERIMENTAL PARA LA ELA
La esclerosis lateral amiotrófica, o ELA, es una enfermedad progresiva del sistema nervioso que afecta las células nerviosas en el cerebro y la médula espinal, y causa pérdida del control muscular.
No hay evidencias de que la enfermedad sea contagiosa. En un pequeño porcentaje aparece la ELA “familiar” con un componente genético hereditario. Sin embargo, el 90-95% de casos de ELA son “esporádicos” esto es, no se transmiten a los hijos. La ELA se creía una enfermedad rara.
La ELA a menudo se llama enfermedad de Lou Gehrig, en honor al jugador de béisbol al que se le diagnosticó la enfermedad. Los médicos generalmente no saben por qué ocurre la ELA. Algunos casos son hereditarios. La ELA es una devastadora enfermedad que afecta a las neuronas motoras. En la mayoría de los casos provoca la muerte en un período de dos a cinco años después de su diagnóstico. Casos como el del reconocido cosmólogo Stephen Hawking, que sobrevivio a la enfermedad más de 50 años, son excepcionales.

La ELA Característicamente produce pérdida de fuerza y atrofia muscular que comienza generalmente por una mano o pierna, afectándose posteriormente el resto de extremidades. Es frecuente que los pacientes observen pequeñas contracciones de algunas partes de su musculatura (fasciculaciones) o calambres dolorosos con los movimientos.
La enfermedad puede producir también dificultad para tragar (disfagia), pronunciar algunas palabras (disartria) o respirar con normalidad (disnea). La progresión de la enfermedad es normalmente irregular, es decir, asimétrica (la enfermedad progresa de modo diferente en cada parte del cuerpo). A veces, la progresión es muy lenta, desarrollándose a los largo de los años y teniendo períodos de estabilidad con un variable grado de incapacidad.
En ningún momento se afectan las facultades intelectuales, ni los órganos de los sentidos (oído, vista, gusto u olfato) ni hay afectación de los esfínteres ni de la función sexual.
La enfermedad cursa sin dolor aunque la presencia de calambres y la pérdida de la movilidad y función muscular acarrean cierto malestar. En algunos casos, aparecen síntomas relacionados con alteraciones de la afectividad (lloros, risas inapropiadas o, en general, respuestas emocionales desproporcionadas como reacción a la afectación física).
UN MECANISMO TOXICO CAUSA LA ELA
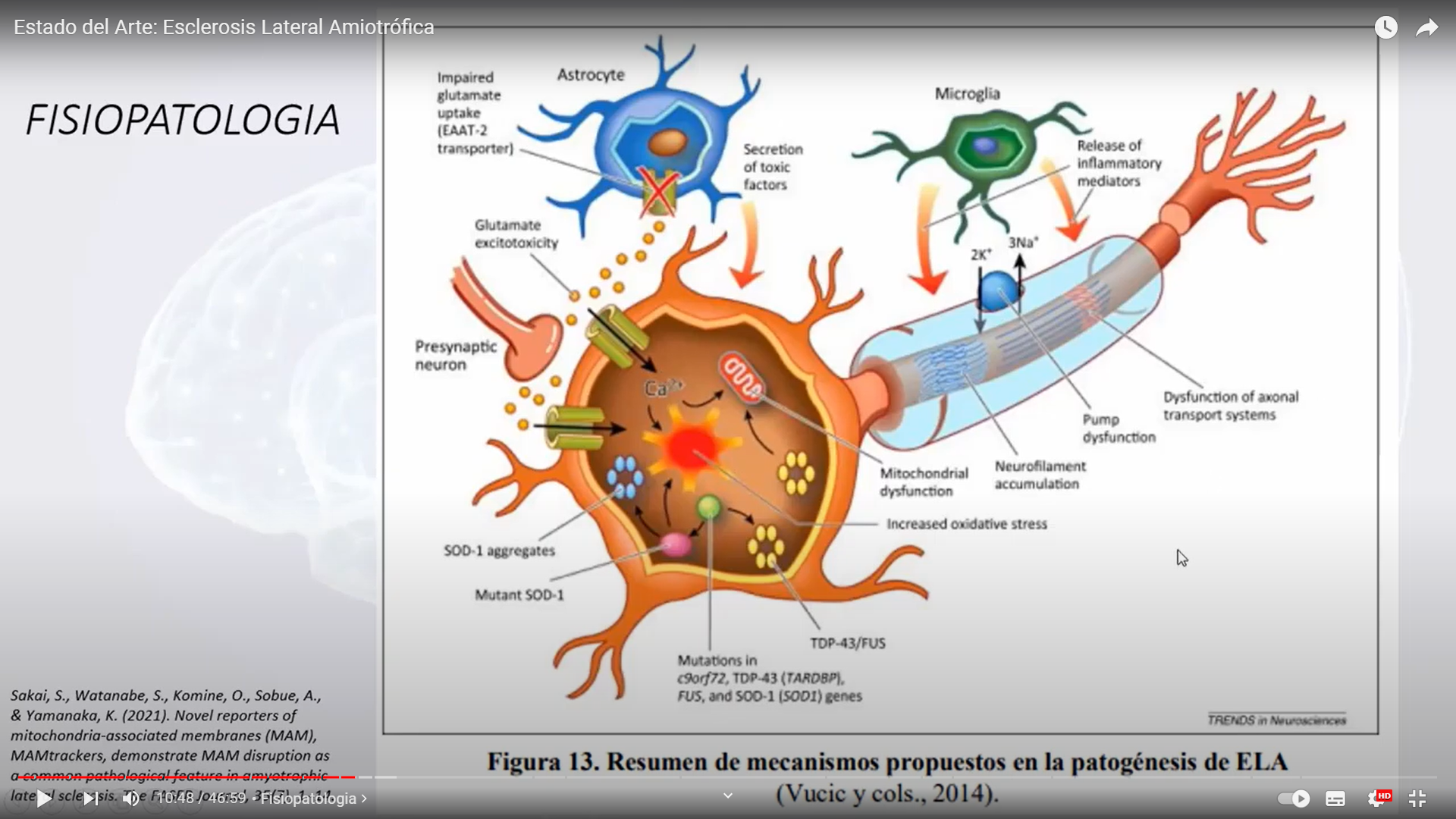
Un nuevo mecanismo tóxico que bloquea cualquier reacción celular que use ácidos nucleicos originaría la muerte de las neuronas motoras en pacientes con ELA.
La esclerosis lateral amiotrófica (ELA), la “enfermedad de Lou Gehrig”, es una enfermedad neurodegenerativa progresiva que afecta a las células nerviosas del cerebro y de la médula espinal. Las neuronas motoras van del cerebro a la médula espinal y de la médula espinal a los músculos de todo el cuerpo.
A-mio-trófica proviene del griego. «A» significa sin o carente. «Mio» se refiere a los músculos, y «trófica» significa alimentación: «Sin alimentación a los músculos». Cuando un músculo no es alimentado, se «atrofia» o se desgasta. «Lateral» identifica las áreas de la médula espinal donde se localizan partes de las células nerviosas que dan señales y controlan los músculos. A medida que esta área se va degenerando, produce la cicatrización o el endurecimiento («esclerosis») en la región.
A medida que las neuronas motoras se van degenerando, dejan de enviar impulsos a las fibras musculares que normalmente resultan en el movimiento muscular. Los primeros síntomas de la ELA a menudo incluyen una mayor debilidad muscular, especialmente en brazos y piernas, en el habla, en la acción de tragar o en la respiración. Cuando los músculos dejan de recibir los mensajes de las neuronas motoras que requieren para funcionar, se empiezan a atrofiar (se vuelven más pequeños). Las extremidades se empiezan a ver más «delgadas», a medida que se atrofia el tejido muscular.
Astas anteriores de la médula espinal con atrofia, degeneración y cromatolisis de neuronas
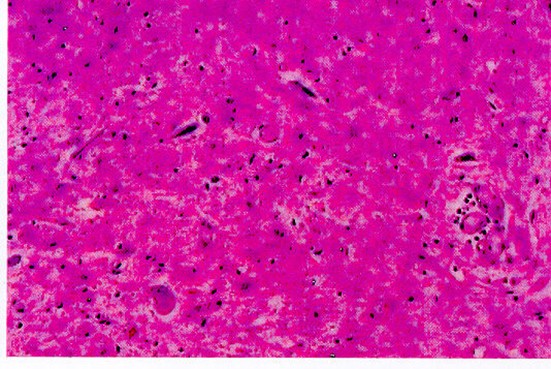
Desmielinización de cordones laterales de medula espinal
FACTORES de riesgo para la ELA
Factor hereditario. Entre el 5 y el 10 % de las personas con esclerosis lateral amiotrófica la heredaron (esclerosis lateral amiotrófica familiar). La mayoría de los hijos de personas con esclerosis lateral amiotrófica familiar tienen un 50 % de probabilidades de desarrollar la enfermedad.
En casi la mitad de estos casos de ELA familiar el origen está en un gen llamado C9ORF72 en el cual, sus mutaciones tienen un papel destacable.
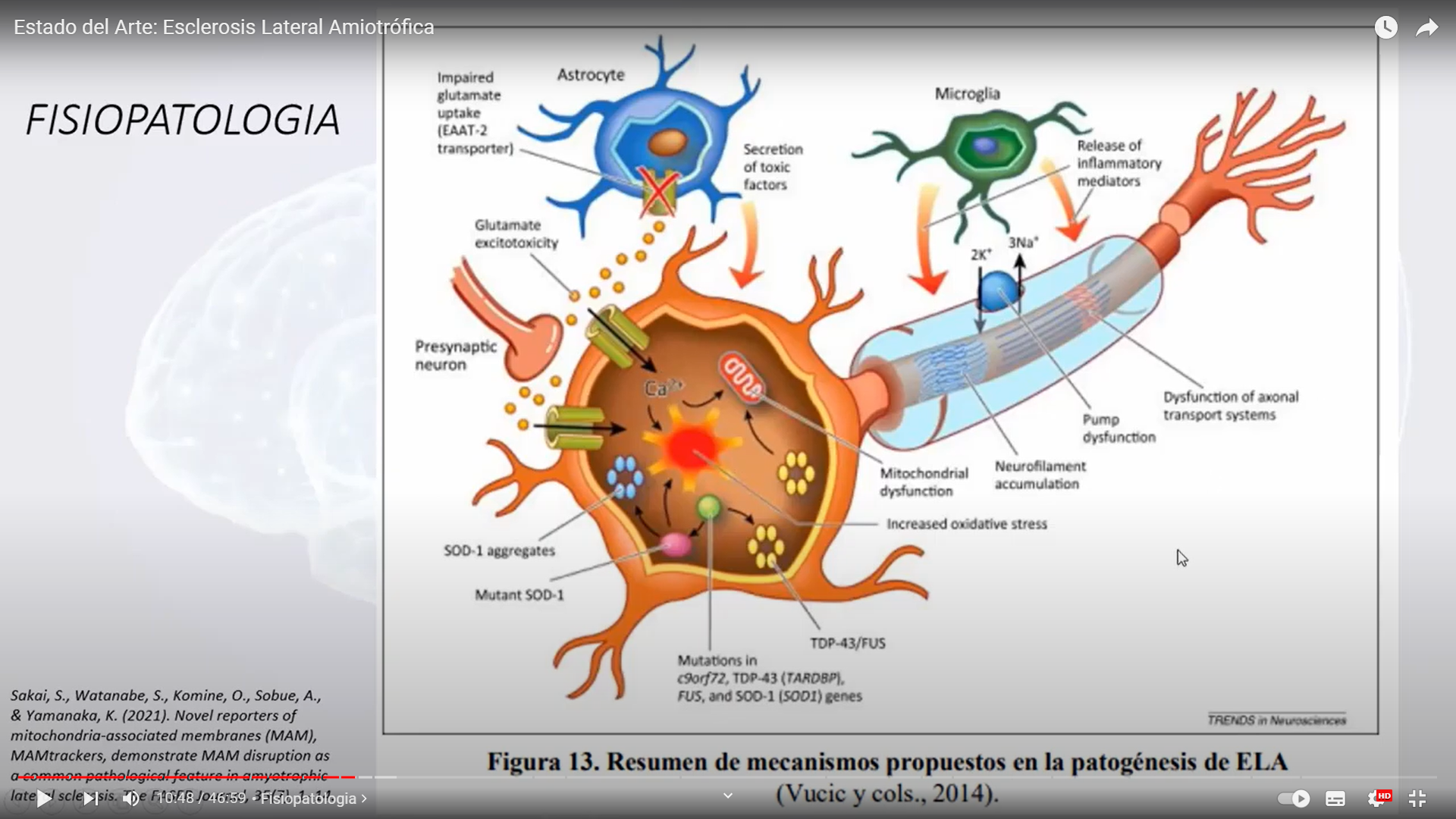
La pregunta clave es por qué las mutaciones en este gen destruyen a las neuronas motoras. Según Óscar Fernández-Capetillo, responsable del Grupo de Inestabilidad Genómica del Centro Nacional de Investigaciones Oncológicas (CNIO), en Madrid, se ha descubierto un mecanismo que explica la toxicidad derivada de mutaciones en el gen C9ORF72.
“A principios de 2011 se descubrió que la mutación más frecuente en pacientes de ELA se encuentra en este gen. Pocos años después, se confirma que esta mutación acaba produciendo unos pequeños péptidos que son tóxicos, pero se desconocía el mecanismo que conducía a ello y que ahora se ha identificado en el CNIO»,
En la mutación del C9ORF72 de los pacientes con ELA existe una pequeña secuencia repetida de ADN que, en personas no enfermas suele ser de unas 8-10 copias, y que en estos pacientes está expandida hasta ciento de veces. “Esta expansión de repeticiones se traducen en proteínas que generan unos pequeños péptidos que son tóxicos, un fenómeno que, por otra parte, se ha observado en otros contextos de la naturaleza humana y para el que hemos desarrollado un modelo que los conecta a todos y explica así estos problemas tan generalizados”.
El mecanismo tóxico identificado y que está asociado a un gen concreto, el C9ORF72, “no se descarta la posibilidad que otras mutaciones relacionadas con ELA estén actuando de manera similar, es decir, bloqueando el ADN y ARN de las neuronas motoras”.
Aprender a aliviar la toxicidad de estos péptidos puede ser útil también para abordar los casos de ELA no asociados a C9ORF72, lo que englobaría a la enfermedad en su conjunto.
“la gran mayoría de las mutaciones halladas en pacientes de ELA son en proteínas que se unen a ARN, y generalmente lo que hacen estas mutaciones es impedir la unión de estas proteínas al ARN».
Además, las células de estos pacientes también tienen problemas muy generales con sus ácidos nucleicos, por lo que «pensamos que, aunque las mutaciones en C9ORF72 afectan a solo una parte de los pacientes de ELA, el mecanismo subyacente a la toxicidad de las neuronas puede que no sea muy diferente, en lo fundamental, a lo que pasa en el resto de los casos de esta enfermedad”.
La ELA podría estar provocada por hongos
Científicos españoles han descrito la posible etiología de la esclerosis lateral amiotrófica (ELA). Proponen que la causa de esta enfermedad se debe a la infección con especies de hongos. El mismo equipo ha presentado evidencias que vinculan las infecciones fúngicas con otras enfermedades neurodegenerativas.
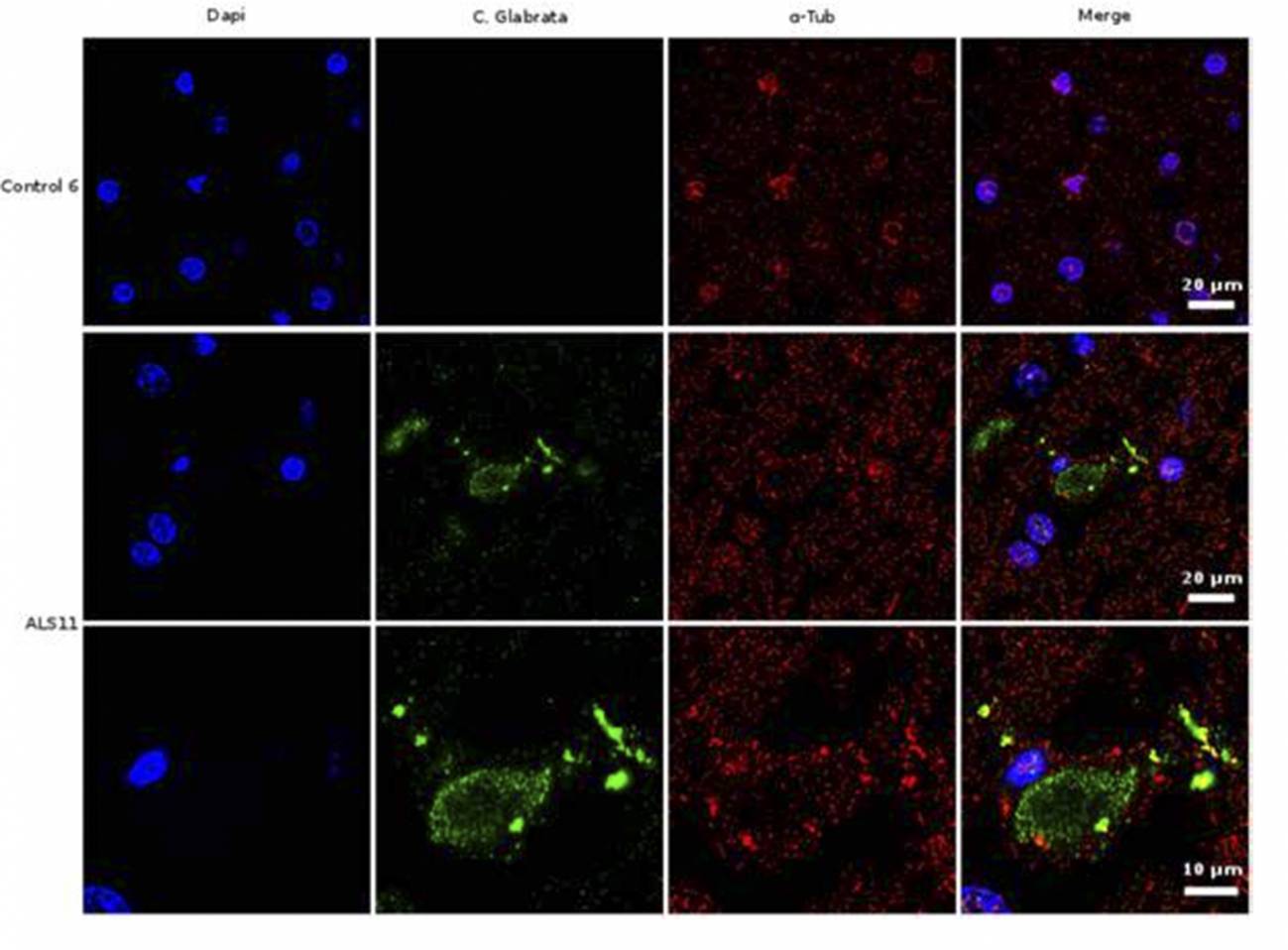
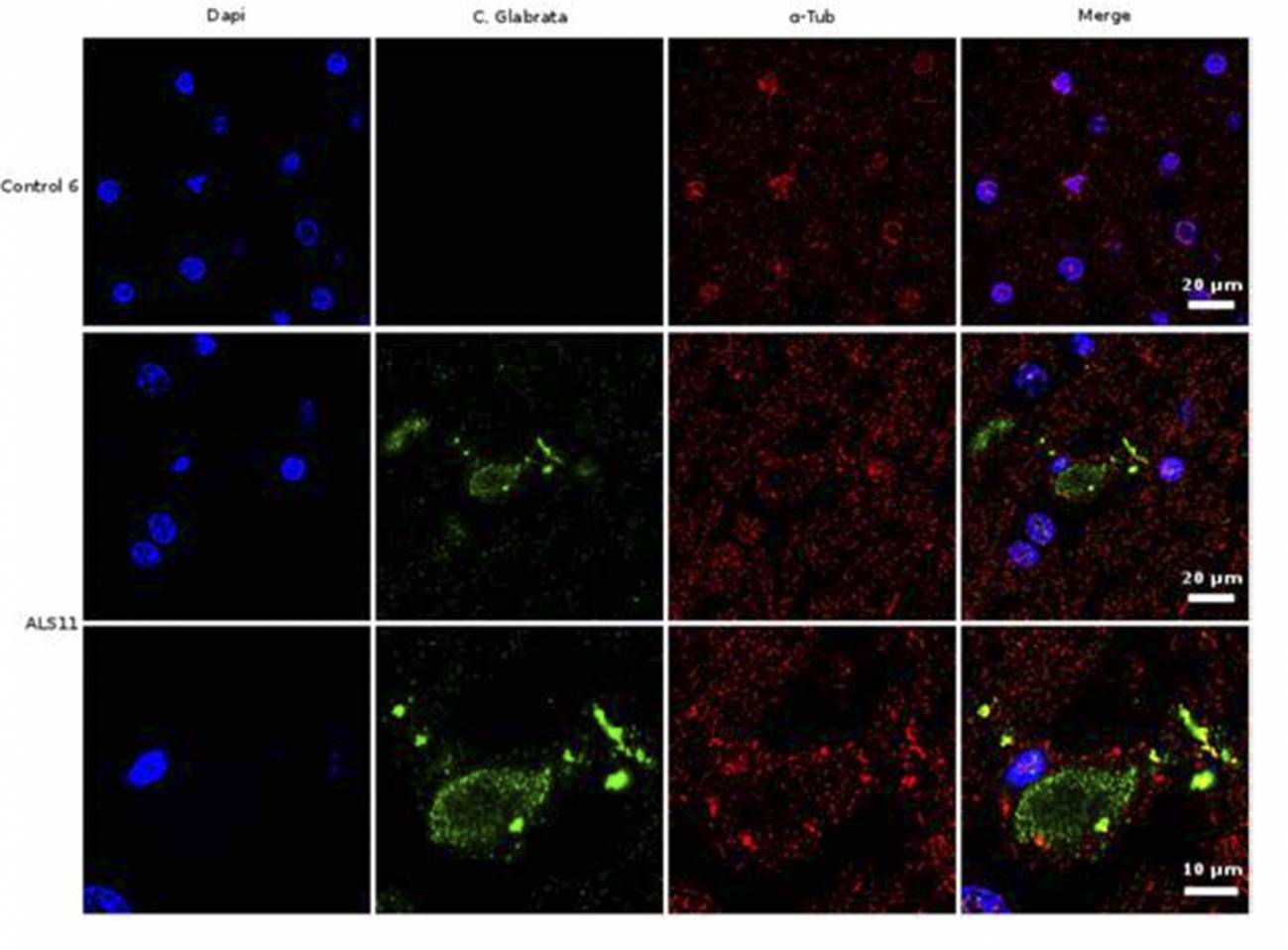
Análisis de inmunohistoquímica de secciones cerebrales de la corteza frontal de una persona control y de un paciente diagnosticado de ELA (ALS11). El núcleo de las células se tiñó con DAPI (azul), mientras que las células de hongo se detectaron con un anticuerpo específico (verde) y la tubulina celular se muestra en rojo. /UAM-ISCIII
El grupo de investigación que lidera Luis Carrasco en el Centro de Biología Molecular Severo Ochoa (CBMSO), centro mixto UAM-CSIC, ha encontrado proteínas fúngicas y DNA de varias especies de hongos en cerebro y líquido cefalorraquídeo de pacientes que padecieron ELA (esclerosis lateral amiotrófica).
Las distintas especies de micosis encontradas (entre otras Candida albicans, Cryptococcus spp. y Malasezzia spp.) podrían ser la causa de dicha enfermedad, según proponen los investigadores en un trabajo publicado en el International Journal of Biological Sciences.
“Nuestros estudios sobre cortes de tejido de cerebro demuestran la existencia de material fúngico y corpúsculos intracelulares, a los que hemos denominado endomicosomas. Estas estructuras fúngicas pueden detectarse mediante inmunofluorescencia, usando anticuerpos que reaccionan específicamente contra proteínas de hongos”, asegura Carrasco.
El equipo de Luis Carrasco, que lleva varios años estudiando la posible etiología de estas enfermedades, ha publicado recientemente distintos trabajos que también vinculan las infecciones fúngicas con el alzhéimer y la esclerosis múltiple.
Además, la diversidad en la evolución y la severidad en los síntomas clínicos observados en este tipo de enfermedades parecen estar relacionadas con el trasfondo genético de cada persona y el estado de su sistema inmune.
Según los autores la comprobación definitiva de que las enfermedades neurodegenerativas están causadas por infecciones con hongos podría obtenerse mediante ensayos clínicos adecuados, los cuales deberían realizarse en hospitales con la colaboración de las compañías farmacéuticas que elaboran compuestos antifúngicos.
En el trabajo también han participado investigadores del Instituto de Salud Carlos III (ISCIII).
La autoridad canadiense ha dado su visto bueno provisional con la condición de que la farmacéutica proporcione más pruebas de que el compuesto AMX0035 funciona El economista, banquero y creador de la fundación que lleva su nombre, Francisco Luzón, enfermo de ELA, durante un curso sobre biología molecular celebrado en la Universidad Internacional Menéndez Pelayo.
Canadá ha aprobado una terapia experimental para la ELA (esclerosis lateral amiotrófica), el trastorno neurológico mortal y paralizante , que agrega una nueva opción de tratamiento para una enfermedad para la que existen pocas terapias efectivas, según recoge un artículo publicado en The New York Times.
La aprobación, la primera en el mundo para el tratamiento, AMX0035,-que se comercializará en Canadá como Albrioza-, se ha realizado con la condición de que la compañía farmacéutica proporcione más pruebas de que el tratamiento funciona. Es probable que sea de gran interés para los pacientes con ELA en Estados Unidos, donde la Administración de Alimentos y Medicamentos (FDA) está evaluando la misma terapia, lo que ha planteado dudas sobre la eficacia del tratamiento.
El artículo de The New York Times explica que una revisión de la FDA de principios de este año encontró que la terapia era segura, pero dijo que no había suficiente evidencia de que fuera efectiva para ayudar a los pacientes a vivir más tiempo o para reducir la velocidad a la que pierden funciones como el control muscular, hablar o respirar sin ayuda. Un comité de asesores independientes de la FDA votó por un estrecho margen en marzo que terapia no estaba lista para su aprobación.
Se había programado que la agencia reguladora de EEUU emitiera una decisión a final este mes, pero finalmente ha extendido la fecha límite hasta el próximo 29 de septiembre, diciendo que necesitaba más tiempo para revisar los análisis adicionales de los datos presentados por la compañía.
Mientras tanto, Calaneet Balas, presidente y director ejecutivo de la Asociación de ELA (ALS Association en inglés), una de las organizaciones de defensa de los pacientes que presionan para obtener la aprobación de la FDA, dijo: «Esperamos que los estadounidenses que viven con ELA intenten acceder a Albrioza en Canadá, tal como hemos escuchado hay personas que intentan comprarlo en Amazon».
Solo hay dos medicamentos para la ELA aprobados en EEUU: riluzol, que puede extender la supervivencia varios meses, y edaravone, que puede retrasar la progresión en aproximadamente un 33%.
Albrioza es una combinación de dos compuestos existentes en forma de un polvo de sabor amargo que se mezcla con agua y se bebe o se ingiere a través de una sonda de alimentación dos veces al día. Es producido por una pequeña empresa de Massachusetts, Amylyx Pharmaceuticals, cuyos fundadores, Justin Klee y Joshua Cohen, concibieron la terapia cuando eran estudiantes en la Universidad de Brown hace menos de una década.
Su idea era que combinar taurursodiol, un suplemento que a veces se usa para regular las enzimas hepáticas, y fenilbutirato de sodio, un medicamento para un trastorno de urea pediátrico, que podría proteger las neuronas al prevenir la disfunción de dos estructuras en las células: las mitocondrias y el retículo endoplásmico.
Los datos que comparan Albrioza con un placebo provienen hasta ahora de un ensayo de fase 2 (que es más reducido que los estudios de fase 3, que generalmente requieren las agencias reguladoras); la información adicional provino de un estudio de extensión de etiqueta abierta que siguió a algunos pacientes después de que finalizó el ensayo, cuando estaban tomando el medicamento a sabiendas. La FDA generalmente requiere dos ensayos clínicos de un medicamento para su aprobación, pero en casos de enfermedad grave con pocos tratamientos disponibles, puede considerar la evidencia de un ensayo clínico más datos de respaldo adicionales.
La autoridad candiense (Health Canadá) dio luz verde a Albrioza bajo la fórmula llamada Aviso de Cumplimiento de Condiciones, que permite la aprobación de medicamentos que parecen prometedores para enfermedades graves, pero tienen evidencia incompleta de que funcionan. La condición central que estableció la agencia es que Amylyx «verifique el beneficio clínico de este medicamento» con datos de un ensayo clínico de fase 3 que está en marcha y se espera que concluya en 2024, según documentos de la agencia enviados a la compañía el viernes. La empresa también debe realizar estudios farmacológicos adicionales y proporcionar informes periódicos de seguridad. Además, la comunicación de la agencia advierte de que «se debe informar a los pacientes
Health Canada dijo en un comunicado: «Los canadienses que viven con ELA tienen opciones limitadas para su tratamiento. Tras una revisión exhaustiva de la información proporcionada por la compañía en su presentación de medicamentos, Health Canada concluyó que los beneficios de Albrioza superan los riesgos cuando se usa según lo previsto».
La FDA tiene un programa similar llamado aprobación acelerada que permite la aprobación condicional de medicamentos con evidencia incompleta de efectividad, pero ese programa también requiere que un compuesto demuestre que se dirige a parte del mecanismo biológico subyacente de una enfermedad. Los expertos han dicho que si Albrioza no obtiene la aprobación estándar de la FDA, es poco probable que cumpla con los criterios de aprobación acelerada porque se sabe muy poco sobre la biología subyacente de la ELA y cómo Albrioza podría abordarla.
El mes pasado, 38 médicos que tratan a pacientes con ELA enviaron una carta a la FDA solicitando la aprobación. La Asociación de ELA dijo que su campaña para la aprobación generó en las últimas semanas más de 6.000 correos electrónicos pidiendo a la agencia que diera luz verde al medicamento.
Un miembro del comité asesor independiente de la FDA, el Dr. G. Caleb Alexander, quien votó en marzo que no había pruebas suficientes de que la terapia funcionara, dijo que sigue pensando que la FDA debería esperar los resultados del ensayo de fase 3 y que «sería un error aprobarlo basándose solo en el ensayo único».
El Dr. Alexander, internista y epidemiólogo de la Escuela de Salud Pública Bloomberg de Johns Hopkins, dijo que había una necesidad desesperada de terapias efectivas para la ELA, pero que para Albrioza, «es desafortunado, pero la magnitud de la necesidad insatisfecha no se corresponde con la calidad de pruebas hasta la fecha.»
Añadió que «la aprobación en Canadá solo podría aumentar aún más la presión que a la que hace frente la FDA para dictaminar favorablemente y aprobar este producto».
Por lo general, es ilegal que los estadounidenses importen medicamentos que no han sido aprobados en los EEUU para uso personal. Pero el sitio web de la FDA enumera algunas excepciones que podrían aplicarse a Albrioza, incluso si el medicamento no tiene problemas de seguridad graves y si es para tratar «una afección grave para la cual no hay un tratamiento efectivo disponible en EEUU».
La Dra. Angela Genge, directora del Centro Global para la Excelencia de la Asociación de ELA en el Instituto Neurológico de Montreal, que recibió honorarios de Amylyx por formar parte de una junta asesora, dijo que los pacientes estadounidenses podrían recibir legalmente Albrioza en Canadá si fuera recetado por un médico canadiense y obtenido de una farmacia canadiense, aunque no serían incluidos para cobertura de seguro bajo el sistema público o privado de Canadá.
En una entrevista, Cohen y Klee se negaron a revelar el precio que Amylyx está considerando para Albrioza, diciendo que todavía se estaba negociando. Dijeron que la terapia estaría disponible en aproximadamente seis semanas para las personas que pagaban de forma privada, pero que llevaría más tiempo, posiblemente meses, para que las personas recibieran cobertura bajo el sistema público de Canadá. Amylyx ya ha proporcionado Albrioza sin costo bajo acuerdos de uso compasivo a 250 pacientes en los EEUU, señalaron.
Hasta el verano pasado, la FDA había recomendado que Amylyx no solicitara la aprobación hasta que el medicamento hubiera completado su ensayo de fase 3, pero en julio, los funcionarios comenzaron a sugerir que Amylyx presentara una solicitud de aprobación utilizando los datos existentes. La presión de los grupos de presión de la ELA se produjo tras la aprobación del nuevo fármaco para el Alzheimer, Aduhelm, que fue muy controverido, pues muchos expertos dijeron que no había datos suficientes de que Aduhelm funcionara.
En el ensayo de fase 2, dos tercios de los 137 participantes recibieron Albrioza y, durante 24 semanas, experimentaron una disminución un 25 % más lenta que los participantes que recibieron placebo: una disminución de 2,32 puntos menos en una escala de 48 puntos en ELA que califica 12 habilidades físicas , incluyendo caminar, hablar, tragar, vestirse, escribir a mano y respirar.
El estudio de extensión de etiqueta abierta involucró a 90 de esos pacientes, incluidos 34 del grupo de placebo, que comenzaron a tomar el medicamento unos siete meses después que los que lo habían recibido desde el principio. Los que recibieron el tratamiento durante más tiempo tuvieron una mediana de unos 6,5 meses más antes de ser hospitalizados, conectados a un ventilador o morir, informó Amylyx. Los investigadores involucrados en el estudio publicaron más datos el mes pasado que sugerían un beneficio adicional.
Amylyx financió la mayor parte de su investigación sobre Albrioza, pero la Asociación de ELA contribuyó con 2.2 millones de dólares, utilizando el dinero recaudado a través del Ice Bucket Challenge 2014 . Amylyx acordó usar las ventas del medicamento para pagar el 150% de la subvención de la asociación para financiar más investigaciones.
Los ensayos clínicos involucraron a pacientes que desarrollaron síntomas dentro de los 18 meses anteriores al ensayo y se vieron afectados en al menos tres regiones del cuerpo, generalmente signos de enfermedad de progreso rápido. La aprobación de Health Canada no marcado restricciones sobre qué pacientes con ELA podían tomar Albrioza, pero los fundadores de Amylyx y el Dr. Genge dijeron que es posible que tales limitaciones las establezca el sistema de cobertura canadiense o los formularios farmacéuticos en algunas provincias de Canadá, concluye el artículo de The New York Times.
Bibliografía:
Mayo Clinic Family Health Book (Libro de Salud Familiar de Mayo Clinic) 5.ª edición
Crawford TO. From enigmatic to problematic. The new molecular genetics of childhood spinal muscular atrophy. Neurology 46: 335-340, 1996.
Ikeda M et al: variable clinical symptoms in familiar amyotrofic lateral sclerosis with a novel point in the Cu/Zn superoxide dismutase gene. Neurology 45: 2038-2041, 1995.
Dr Angela Genge Executive Director, Clinical Research Unit, Montreal Neurological Institute and Hospital : «ALS Drug Development Programs 2021: Overview of the international ALS drug development programs with the integration of Biomarkers in Early Phase development programs»
TERAPIA EXPERIMENTAL PARA LA ELA
La esclerosis lateral amiotrófica, o ELA, es una enfermedad progresiva del sistema nervioso que afecta las células nerviosas en el cerebro y la médula espinal, y causa pérdida del control muscular.
No hay evidencias de que la enfermedad sea contagiosa. En un pequeño porcentaje aparece la ELA “familiar” con un componente genético hereditario. Sin embargo, el 90-95% de casos de ELA son “esporádicos” esto es, no se transmiten a los hijos. La ELA se creía una enfermedad rara.
La ELA a menudo se llama enfermedad de Lou Gehrig, en honor al jugador de béisbol al que se le diagnosticó la enfermedad. Los médicos generalmente no saben por qué ocurre la ELA. Algunos casos son hereditarios. La ELA es una devastadora enfermedad que afecta a las neuronas motoras. En la mayoría de los casos provoca la muerte en un período de dos a cinco años después de su diagnóstico. Casos como el del reconocido cosmólogo Stephen Hawking, que sobrevivio a la enfermedad más de 50 años, son excepcionales.

La ELA Característicamente produce pérdida de fuerza y atrofia muscular que comienza generalmente por una mano o pierna, afectándose posteriormente el resto de extremidades. Es frecuente que los pacientes observen pequeñas contracciones de algunas partes de su musculatura (fasciculaciones) o calambres dolorosos con los movimientos.
La enfermedad puede producir también dificultad para tragar (disfagia), pronunciar algunas palabras (disartria) o respirar con normalidad (disnea). La progresión de la enfermedad es normalmente irregular, es decir, asimétrica (la enfermedad progresa de modo diferente en cada parte del cuerpo). A veces, la progresión es muy lenta, desarrollándose a los largo de los años y teniendo períodos de estabilidad con un variable grado de incapacidad.
En ningún momento se afectan las facultades intelectuales, ni los órganos de los sentidos (oído, vista, gusto u olfato) ni hay afectación de los esfínteres ni de la función sexual.
La enfermedad cursa sin dolor aunque la presencia de calambres y la pérdida de la movilidad y función muscular acarrean cierto malestar. En algunos casos, aparecen síntomas relacionados con alteraciones de la afectividad (lloros, risas inapropiadas o, en general, respuestas emocionales desproporcionadas como reacción a la afectación física).
UN MECANISMO TOXICO CAUSA LA ELA
Un nuevo mecanismo tóxico que bloquea cualquier reacción celular que use ácidos nucleicos originaría la muerte de las neuronas motoras en pacientes con ELA.
La esclerosis lateral amiotrófica (ELA), la “enfermedad de Lou Gehrig”, es una enfermedad neurodegenerativa progresiva que afecta a las células nerviosas del cerebro y de la médula espinal. Las neuronas motoras van del cerebro a la médula espinal y de la médula espinal a los músculos de todo el cuerpo.
A-mio-trófica proviene del griego. «A» significa sin o carente. «Mio» se refiere a los músculos, y «trófica» significa alimentación: «Sin alimentación a los músculos». Cuando un músculo no es alimentado, se «atrofia» o se desgasta. «Lateral» identifica las áreas de la médula espinal donde se localizan partes de las células nerviosas que dan señales y controlan los músculos. A medida que esta área se va degenerando, produce la cicatrización o el endurecimiento («esclerosis») en la región.
A medida que las neuronas motoras se van degenerando, dejan de enviar impulsos a las fibras musculares que normalmente resultan en el movimiento muscular. Los primeros síntomas de la ELA a menudo incluyen una mayor debilidad muscular, especialmente en brazos y piernas, en el habla, en la acción de tragar o en la respiración. Cuando los músculos dejan de recibir los mensajes de las neuronas motoras que requieren para funcionar, se empiezan a atrofiar (se vuelven más pequeños). Las extremidades se empiezan a ver más «delgadas», a medida que se atrofia el tejido muscular.
Astas anteriores de la médula espinal con atrofia, degeneración y cromatolisis de neuronas
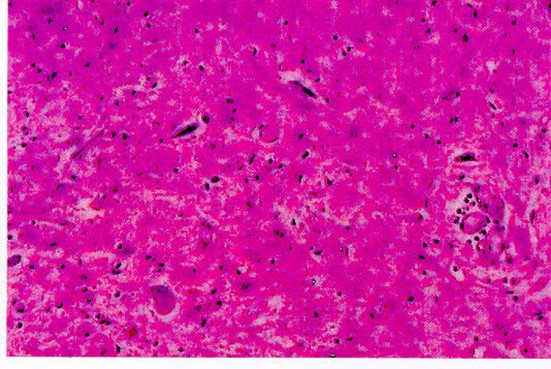
Desmielinización de cordones laterales de medula espinal
FACTORES de riesgo para la ELA
Factor hereditario. Entre el 5 y el 10 % de las personas con esclerosis lateral amiotrófica la heredaron (esclerosis lateral amiotrófica familiar). La mayoría de los hijos de personas con esclerosis lateral amiotrófica familiar tienen un 50 % de probabilidades de desarrollar la enfermedad.
En casi la mitad de estos casos de ELA familiar el origen está en un gen llamado C9ORF72 en el cual, sus mutaciones tienen un papel destacable.
La pregunta clave es por qué las mutaciones en este gen destruyen a las neuronas motoras. Según Óscar Fernández-Capetillo, responsable del Grupo de Inestabilidad Genómica del Centro Nacional de Investigaciones Oncológicas (CNIO), en Madrid, se ha descubierto un mecanismo que explica la toxicidad derivada de mutaciones en el gen C9ORF72.
“A principios de 2011 se descubrió que la mutación más frecuente en pacientes de ELA se encuentra en este gen. Pocos años después, se confirma que esta mutación acaba produciendo unos pequeños péptidos que son tóxicos, pero se desconocía el mecanismo que conducía a ello y que ahora se ha identificado en el CNIO»,
En la mutación del C9ORF72 de los pacientes con ELA existe una pequeña secuencia repetida de ADN que, en personas no enfermas suele ser de unas 8-10 copias, y que en estos pacientes está expandida hasta ciento de veces. “Esta expansión de repeticiones se traducen en proteínas que generan unos pequeños péptidos que son tóxicos, un fenómeno que, por otra parte, se ha observado en otros contextos de la naturaleza humana y para el que hemos desarrollado un modelo que los conecta a todos y explica así estos problemas tan generalizados”.
El mecanismo tóxico identificado y que está asociado a un gen concreto, el C9ORF72, “no se descarta la posibilidad que otras mutaciones relacionadas con ELA estén actuando de manera similar, es decir, bloqueando el ADN y ARN de las neuronas motoras”.
Aprender a aliviar la toxicidad de estos péptidos puede ser útil también para abordar los casos de ELA no asociados a C9ORF72, lo que englobaría a la enfermedad en su conjunto.
“la gran mayoría de las mutaciones halladas en pacientes de ELA son en proteínas que se unen a ARN, y generalmente lo que hacen estas mutaciones es impedir la unión de estas proteínas al ARN».
Además, las células de estos pacientes también tienen problemas muy generales con sus ácidos nucleicos, por lo que «pensamos que, aunque las mutaciones en C9ORF72 afectan a solo una parte de los pacientes de ELA, el mecanismo subyacente a la toxicidad de las neuronas puede que no sea muy diferente, en lo fundamental, a lo que pasa en el resto de los casos de esta enfermedad”.
La ELA podría estar provocada por hongos
Científicos españoles han descrito la posible etiología de la esclerosis lateral amiotrófica (ELA). Proponen que la causa de esta enfermedad se debe a la infección con especies de hongos. El mismo equipo ha presentado evidencias que vinculan las infecciones fúngicas con otras enfermedades neurodegenerativas.
Análisis de inmunohistoquímica de secciones cerebrales de la corteza frontal de una persona control y de un paciente diagnosticado de ELA (ALS11). El núcleo de las células se tiñó con DAPI (azul), mientras que las células de hongo se detectaron con un anticuerpo específico (verde) y la tubulina celular se muestra en rojo. /UAM-ISCIII
El grupo de investigación que lidera Luis Carrasco en el Centro de Biología Molecular Severo Ochoa (CBMSO), centro mixto UAM-CSIC, ha encontrado proteínas fúngicas y DNA de varias especies de hongos en cerebro y líquido cefalorraquídeo de pacientes que padecieron ELA (esclerosis lateral amiotrófica).
Las distintas especies de micosis encontradas (entre otras Candida albicans, Cryptococcus spp. y Malasezzia spp.) podrían ser la causa de dicha enfermedad, según proponen los investigadores en un trabajo publicado en el International Journal of Biological Sciences.
“Nuestros estudios sobre cortes de tejido de cerebro demuestran la existencia de material fúngico y corpúsculos intracelulares, a los que hemos denominado endomicosomas. Estas estructuras fúngicas pueden detectarse mediante inmunofluorescencia, usando anticuerpos que reaccionan específicamente contra proteínas de hongos”, asegura Carrasco.
El equipo de Luis Carrasco, que lleva varios años estudiando la posible etiología de estas enfermedades, ha publicado recientemente distintos trabajos que también vinculan las infecciones fúngicas con el alzhéimer y la esclerosis múltiple.
Además, la diversidad en la evolución y la severidad en los síntomas clínicos observados en este tipo de enfermedades parecen estar relacionadas con el trasfondo genético de cada persona y el estado de su sistema inmune.
Según los autores la comprobación definitiva de que las enfermedades neurodegenerativas están causadas por infecciones con hongos podría obtenerse mediante ensayos clínicos adecuados, los cuales deberían realizarse en hospitales con la colaboración de las compañías farmacéuticas que elaboran compuestos antifúngicos.
En el trabajo también han participado investigadores del Instituto de Salud Carlos III (ISCIII).
La autoridad canadiense ha dado su visto bueno provisional con la condición de que la farmacéutica proporcione más pruebas de que el compuesto AMX0035 funciona El economista, banquero y creador de la fundación que lleva su nombre, Francisco Luzón, enfermo de ELA, durante un curso sobre biología molecular celebrado en la Universidad Internacional Menéndez Pelayo.
Canadá ha aprobado una terapia experimental para la ELA (esclerosis lateral amiotrófica), el trastorno neurológico mortal y paralizante , que agrega una nueva opción de tratamiento para una enfermedad para la que existen pocas terapias efectivas, según recoge un artículo publicado en The New York Times.
La aprobación, la primera en el mundo para el tratamiento, AMX0035,-que se comercializará en Canadá como Albrioza-, se ha realizado con la condición de que la compañía farmacéutica proporcione más pruebas de que el tratamiento funciona. Es probable que sea de gran interés para los pacientes con ELA en Estados Unidos, donde la Administración de Alimentos y Medicamentos (FDA) está evaluando la misma terapia, lo que ha planteado dudas sobre la eficacia del tratamiento.
El artículo de The New York Times explica que una revisión de la FDA de principios de este año encontró que la terapia era segura, pero dijo que no había suficiente evidencia de que fuera efectiva para ayudar a los pacientes a vivir más tiempo o para reducir la velocidad a la que pierden funciones como el control muscular, hablar o respirar sin ayuda. Un comité de asesores independientes de la FDA votó por un estrecho margen en marzo que terapia no estaba lista para su aprobación.
Se había programado que la agencia reguladora de EEUU emitiera una decisión a final este mes, pero finalmente ha extendido la fecha límite hasta el próximo 29 de septiembre, diciendo que necesitaba más tiempo para revisar los análisis adicionales de los datos presentados por la compañía.
Mientras tanto, Calaneet Balas, presidente y director ejecutivo de la Asociación de ELA (ALS Association en inglés), una de las organizaciones de defensa de los pacientes que presionan para obtener la aprobación de la FDA, dijo: «Esperamos que los estadounidenses que viven con ELA intenten acceder a Albrioza en Canadá, tal como hemos escuchado hay personas que intentan comprarlo en Amazon».
Solo hay dos medicamentos para la ELA aprobados en EEUU: riluzol, que puede extender la supervivencia varios meses, y edaravone, que puede retrasar la progresión en aproximadamente un 33%.
Albrioza es una combinación de dos compuestos existentes en forma de un polvo de sabor amargo que se mezcla con agua y se bebe o se ingiere a través de una sonda de alimentación dos veces al día. Es producido por una pequeña empresa de Massachusetts, Amylyx Pharmaceuticals, cuyos fundadores, Justin Klee y Joshua Cohen, concibieron la terapia cuando eran estudiantes en la Universidad de Brown hace menos de una década.
Su idea era que combinar taurursodiol, un suplemento que a veces se usa para regular las enzimas hepáticas, y fenilbutirato de sodio, un medicamento para un trastorno de urea pediátrico, que podría proteger las neuronas al prevenir la disfunción de dos estructuras en las células: las mitocondrias y el retículo endoplásmico.
Los datos que comparan Albrioza con un placebo provienen hasta ahora de un ensayo de fase 2 (que es más reducido que los estudios de fase 3, que generalmente requieren las agencias reguladoras); la información adicional provino de un estudio de extensión de etiqueta abierta que siguió a algunos pacientes después de que finalizó el ensayo, cuando estaban tomando el medicamento a sabiendas. La FDA generalmente requiere dos ensayos clínicos de un medicamento para su aprobación, pero en casos de enfermedad grave con pocos tratamientos disponibles, puede considerar la evidencia de un ensayo clínico más datos de respaldo adicionales.
La autoridad candiense (Health Canadá) dio luz verde a Albrioza bajo la fórmula llamada Aviso de Cumplimiento de Condiciones, que permite la aprobación de medicamentos que parecen prometedores para enfermedades graves, pero tienen evidencia incompleta de que funcionan. La condición central que estableció la agencia es que Amylyx «verifique el beneficio clínico de este medicamento» con datos de un ensayo clínico de fase 3 que está en marcha y se espera que concluya en 2024, según documentos de la agencia enviados a la compañía el viernes. La empresa también debe realizar estudios farmacológicos adicionales y proporcionar informes periódicos de seguridad. Además, la comunicación de la agencia advierte de que «se debe informar a los pacientes
Health Canada dijo en un comunicado: «Los canadienses que viven con ELA tienen opciones limitadas para su tratamiento. Tras una revisión exhaustiva de la información proporcionada por la compañía en su presentación de medicamentos, Health Canada concluyó que los beneficios de Albrioza superan los riesgos cuando se usa según lo previsto».
La FDA tiene un programa similar llamado aprobación acelerada que permite la aprobación condicional de medicamentos con evidencia incompleta de efectividad, pero ese programa también requiere que un compuesto demuestre que se dirige a parte del mecanismo biológico subyacente de una enfermedad. Los expertos han dicho que si Albrioza no obtiene la aprobación estándar de la FDA, es poco probable que cumpla con los criterios de aprobación acelerada porque se sabe muy poco sobre la biología subyacente de la ELA y cómo Albrioza podría abordarla.
El mes pasado, 38 médicos que tratan a pacientes con ELA enviaron una carta a la FDA solicitando la aprobación. La Asociación de ELA dijo que su campaña para la aprobación generó en las últimas semanas más de 6.000 correos electrónicos pidiendo a la agencia que diera luz verde al medicamento.
Un miembro del comité asesor independiente de la FDA, el Dr. G. Caleb Alexander, quien votó en marzo que no había pruebas suficientes de que la terapia funcionara, dijo que sigue pensando que la FDA debería esperar los resultados del ensayo de fase 3 y que «sería un error aprobarlo basándose solo en el ensayo único».
El Dr. Alexander, internista y epidemiólogo de la Escuela de Salud Pública Bloomberg de Johns Hopkins, dijo que había una necesidad desesperada de terapias efectivas para la ELA, pero que para Albrioza, «es desafortunado, pero la magnitud de la necesidad insatisfecha no se corresponde con la calidad de pruebas hasta la fecha.»
Añadió que «la aprobación en Canadá solo podría aumentar aún más la presión que a la que hace frente la FDA para dictaminar favorablemente y aprobar este producto».
Por lo general, es ilegal que los estadounidenses importen medicamentos que no han sido aprobados en los EEUU para uso personal. Pero el sitio web de la FDA enumera algunas excepciones que podrían aplicarse a Albrioza, incluso si el medicamento no tiene problemas de seguridad graves y si es para tratar «una afección grave para la cual no hay un tratamiento efectivo disponible en EEUU».
La Dra. Angela Genge, directora del Centro Global para la Excelencia de la Asociación de ELA en el Instituto Neurológico de Montreal, que recibió honorarios de Amylyx por formar parte de una junta asesora, dijo que los pacientes estadounidenses podrían recibir legalmente Albrioza en Canadá si fuera recetado por un médico canadiense y obtenido de una farmacia canadiense, aunque no serían incluidos para cobertura de seguro bajo el sistema público o privado de Canadá.
En una entrevista, Cohen y Klee se negaron a revelar el precio que Amylyx está considerando para Albrioza, diciendo que todavía se estaba negociando. Dijeron que la terapia estaría disponible en aproximadamente seis semanas para las personas que pagaban de forma privada, pero que llevaría más tiempo, posiblemente meses, para que las personas recibieran cobertura bajo el sistema público de Canadá. Amylyx ya ha proporcionado Albrioza sin costo bajo acuerdos de uso compasivo a 250 pacientes en los EEUU, señalaron.
Hasta el verano pasado, la FDA había recomendado que Amylyx no solicitara la aprobación hasta que el medicamento hubiera completado su ensayo de fase 3, pero en julio, los funcionarios comenzaron a sugerir que Amylyx presentara una solicitud de aprobación utilizando los datos existentes. La presión de los grupos de presión de la ELA se produjo tras la aprobación del nuevo fármaco para el Alzheimer, Aduhelm, que fue muy controverido, pues muchos expertos dijeron que no había datos suficientes de que Aduhelm funcionara.
En el ensayo de fase 2, dos tercios de los 137 participantes recibieron Albrioza y, durante 24 semanas, experimentaron una disminución un 25 % más lenta que los participantes que recibieron placebo: una disminución de 2,32 puntos menos en una escala de 48 puntos en ELA que califica 12 habilidades físicas , incluyendo caminar, hablar, tragar, vestirse, escribir a mano y respirar.
El estudio de extensión de etiqueta abierta involucró a 90 de esos pacientes, incluidos 34 del grupo de placebo, que comenzaron a tomar el medicamento unos siete meses después que los que lo habían recibido desde el principio. Los que recibieron el tratamiento durante más tiempo tuvieron una mediana de unos 6,5 meses más antes de ser hospitalizados, conectados a un ventilador o morir, informó Amylyx. Los investigadores involucrados en el estudio publicaron más datos el mes pasado que sugerían un beneficio adicional.
Amylyx financió la mayor parte de su investigación sobre Albrioza, pero la Asociación de ELA contribuyó con 2.2 millones de dólares, utilizando el dinero recaudado a través del Ice Bucket Challenge 2014 . Amylyx acordó usar las ventas del medicamento para pagar el 150% de la subvención de la asociación para financiar más investigaciones.
Los ensayos clínicos involucraron a pacientes que desarrollaron síntomas dentro de los 18 meses anteriores al ensayo y se vieron afectados en al menos tres regiones del cuerpo, generalmente signos de enfermedad de progreso rápido. La aprobación de Health Canada no marcado restricciones sobre qué pacientes con ELA podían tomar Albrioza, pero los fundadores de Amylyx y el Dr. Genge dijeron que es posible que tales limitaciones las establezca el sistema de cobertura canadiense o los formularios farmacéuticos en algunas provincias de Canadá, concluye el artículo de The New York Times.
Bibliografía:
Mayo Clinic Family Health Book (Libro de Salud Familiar de Mayo Clinic) 5.ª edición
Crawford TO. From enigmatic to problematic. The new molecular genetics of childhood spinal muscular atrophy. Neurology 46: 335-340, 1996.
Ikeda M et al: variable clinical symptoms in familiar amyotrofic lateral sclerosis with a novel point in the Cu/Zn superoxide dismutase gene. Neurology 45: 2038-2041, 1995.
Dr Angela Genge Executive Director, Clinical Research Unit, Montreal Neurological Institute and Hospital : «ALS Drug Development Programs 2021: Overview of the international ALS drug development programs with the integration of Biomarkers in Early Phase development programs»