El trastorno del espectro autista (TEA) abarca un grupo de trastornos multifactoriales del neurodesarrollo caracterizados por una comunicación e interacción social deteriorada y por comportamientos repetitivos y estereotipados. Múltiples estudios han revelado que en el TEA existen disfunciones sinápticas, en la cual la morfología y función neuronal son sustratos importantes en esta patogenia. En esta revisión comentamos los datos disponibles a nivel de anormalidades neuronales en el TEA, enfatizando la morfología de las dendritas, espinas dendríticas y citoesquelo de actina. Las dendritas y espinas dendríticas, ricas en actina, forman la parte postsináptica de la mayoría de las sinapsis excitadoras. En el TEA, los datos obtenidos apuntan a una desregulación en el crecimiento y desarrollo dendrítico, así como una alteración en la densidad de las espinas dendríticas. Lo anterior, se ve acompañado de alteraciones en la remodelación y composición del citoesqueleto neuronal. Para comprender mejor la fisiopatología del TEA, es necesario mayor información sobre cómo los cambios morfofuncionales de los actores que participan en la sinapsis impactan en los circuitos y el comportamiento.

El trastorno del espectro autista (TEA) se caracteriza por deficiencias en la interacción social, el lenguaje, el comportamiento y las funciones cognitivas (APA, 2013). El fenotipo del autismo no es idéntico en las personas afectadas. La heterogeneidad de la progresión de la enfermedad, la gravedad de sus síntomas y los diversos trastornos asociados comórbidos llevan a creer en sustratos neurológicos variables de la enfermedad. Actualmente, existe un debate en curso acerca de cuán permanentes son las alteraciones neuromorfológicas en el cerebro y si existe una plasticidad reversible de tales cambios. Diversas investigaciones se han dedicado a los períodos críticos en el desarrollo del cerebro en el cual son sensibles a factores ambientales
Los tratornos organicos, explican con mas rotundidez los hallazgos funcionales. El trabajo que sigue, demuestra que una microglía, impide la poda neuronal que caracteriza a los TEA. Los autofagosomas, unas vesículas citoplasmáticas que sirven para capturar y transportar componentes celulares a los lisosomas y allí destruirlos.
Las personas con TEA tienen más sinapsis, por una poda limitada y esa poda se produce por autofagia. La microglía se come las sinapsis no funcionales. Otra evidencia de conexión sería genética ya que en el TEA se han visto mutaciones de variación en el número de copias en genes que codifican proteínas implicadas en las vías autofágicas.
Una teoría sobre el origen del TEA propone que se debe a una alteración de la autofagia por una hiperactivación de mTOR.
La mTOR es necesaria para la poda de espinas en el desarrollo, y que la activación de la autofagia neuronal corrige la patología sináptica y los déficits de comportamiento social en modelos animales de TEA con mTOR hiperactivo
El déficit en la poda de espinas se correlaciona con la hiperactivación de mTOR y el deterioro de la autofagia.
mTOR, son las siglas de una proteína que corresponden a «mammalian target of rapamycin» o «diana de rapamicina en células de mamífero». mTOR interviene en importantes procesos celulares, incluyendo el crecimiento, la proliferación, la motilidad, la supervivencia, la síntesis de proteínas, la transcripción y la autofagia.
La autofagia regulada por mTOR es necesaria para la poda de espinas en el desarrollo, y que la activación de la autofagia neuronal corrige la patología sináptica y los déficits de comportamiento social en modelos animales de TEA con mTOR hiperactivo.
La activación de mTOR inhibe la autofagia en un paso temprano de la formación de autofagosomas.
En 1972, Suren Sehgal identificó una pequeña molécula procedente de la bacteria del suelo Streptomyces hygroscopicus, que purificó y vio que poseía una potente actividad antifúngica. La denominó rapamicina y las pruebas posteriores revelaron que también tenía una potente actividad inmunosupresora y citostática contra el cáncer. Se utiliza en la actualidad para los trasplantados de riñón.
Aunque los TEA presentan una gran heterogeneidad genética y clínica, varios síndromes de TEA están causados por mutaciones en genes que codifican proteínas que inhiben mTOR; es decir, esas mutaciones hacen perder la inhibición y generan una hiperactividad de mTOR. Entre estos genes están Tsc1/Tsc2, NF1 y ptPten.
La mTOR es un regulador clave de la síntesis de proteínas sinápticas, y las aberraciones en la señalización de la mTOR se han vinculado a las anomalías sinápticas y neuroanatómicas que se observan en los TEA sindrómicos (por ejemplo, la esclerosis tuberosa) e idiopáticos. Entre las proteínas implicadas y que también se han relacionado con el TEA están SHANK3, FMRP y los receptores de glutamato mGluR1/5.
La mTOR está implicada en la formación de autofagosomas, unas vesículas citoplasmáticas que sirven para capturar y transportar componentes celulares a los lisosomas y allí destruirlos. Este proceso, conocido como autofagia o macroautofagia, es importante para eliminar los orgánulos dañados y degradar las proteínas de larga vida o propensas a formar agregados.
Menos poda implica más espinas y eso es lo que se ve en las personas con autismo.
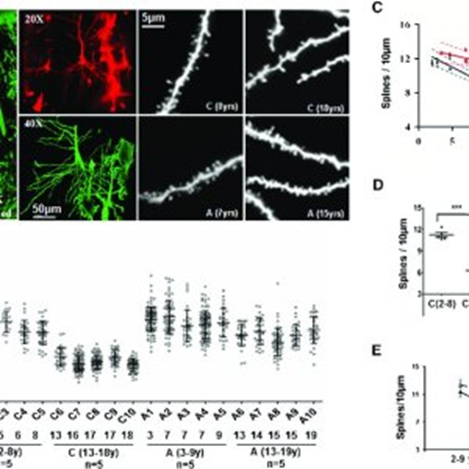
Otras investigaciones de los mecanismos implicados realizadas por Tang et al. (2014), sugieren que el excedente de espinas excitatorias observado podría ser el resultado de una alteración en la autofagia impulsada por mTOR y una poda de espinas deficiente, estableciendo así un vínculo entre una anomalía de la señalización neuronal en el TEA y las alteraciones sinápticas en estos trastornos.
En los ratones Tsc2 +/- , un modelo animal de TEA, y en los que mTOR está hiperactivo de forma constitutiva, Tang et al. (2014) observaron defectos en la poda postnatal de espinas, bloqueo de la autofagia y comportamientos sociales anómalos similares a los del TEA, lo que encaja con todo lo que hemos visto antes. Un resultado impactante es que la rapamicina, el inhibidor de mTOR, corrigió los comportamientos similares al TEA y los defectos de poda sináptica en estos ratones Tsc2 +/-.
Los resultados de este artículo sugieren que la autofagia regulada por mTOR es necesaria para la poda de espinas en el desarrollo, y que la activación de la autofagia neuronal corrige la patología sináptica y los déficits de comportamiento social en modelos animales de TEA con mTOR hiperactivo.
Para determinar la presencia de alteraciones sinápticas en los mutantes Tsc2+/-, Pagani et al., (2021) midieron la densidad de espinas dendríticas en el córtex insular, una región relevante para la disfunción social en el autismo, y descubrieron que los ratones Tsc2+/- presentan una mayor densidad de espinas en comparación con sus compañeros de camada que servían de control. A continuación, usaron resonancia magnética funcional para mapear la conectividad de todo el cerebro en mutantes Tsc2+/- juveniles (P28). El uso de ratones prepúberes permite identificar un patrón de conectividad no afectado por la remodelación sináptica y de circuitos que se produce con la pubertad, y por tanto sería más indicativa de los circuitos neuronales que caracterizan los síntomas tempranos en el TEA.
Pagani et al. (2021) trataron los ratones Tsc2+/- y los ratones control con rapamicina, el inhibidor de mTOR, durante su cuarta semana postnatal.
La cuantificación de la densidad de espinas dendríticas reveló que el tratamiento con rapamicina redujo la densidad de espinas en los ratones Tsc2+/- hasta retornar a niveles comparables a los de los ratones Tsc2+/+ de control (P < 0,001). Sorprendentemente, la resonancia magnética funcional de los ratones Tsc2+/- tratados con rapamicina también reveló un rescate completo del fenotipo de hiperconectividad, lo que implica una marcada reducción de la conectividad funcional de largo alcance en las mismas regiones de la corteza y el estriado que se caracterizan por la hiperconectividad en los mutantes Tsc2+/- tratados con vehículo. En consonancia con esto, la rapamicina también rescató la conectividad de la red por defecto y la red de saliencia en los ratones Tsc2+/-. Estos resultados corroboran la especificidad del mecanismo, apoyan un vínculo causal entre la patología sináptica dependiente de mTOR y la hiperconectividad en los ratones Tsc2+/- y plantean una posibilidad de terapia farmacológica, el uso de la rapamicina.
Bibliografia
Pagani M, Barsotti N, Bertero A, Trakoshis S, Ulysse L, Locarno A, Miseviciute I, De Felice A, Canella C, Supekar K, Galbusera A, Menon V, Tonini R, Deco G, Lombardo MV, Pasqualetti M, Gozzi A (2021) mTOR-related synaptic pathology causes autism spectrum disorder-associated functional hyperconnectivity. Nat Commun 12(1): 6084.
Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS, Kanter E, Castagna C, Yamamoto A, Yue Z, Arancio O, Peterson BS, Champagne F, Dwork AJ, Goldman J, Sulzer D (2014) Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83: 1131–1143.