EPIGENETICA
Cada dia , pero cada dia un nuevo conocimiento científico, modifica a lo anterior.
Y no es posible ignoraralo porque en ellos esta la vida.
Shinya Yamanaka 2006, John B. Gurdon reprogramación celular
LA APARICION DE LA EPIGENETICA, ES UNA REVOLUCION COMO LO SON CADA DIA, LAS NUEVAS APARICIONES.
Las dificultades del conocimiento vienen porporcionada, porque los nuevos hallazgos, desmontan totalmente lo anterior.
Esto es una copia de una conferencia Hispano Americana sobre epigenetica
FÉLIX GOÑI catedrático de bioquímica y biología molecular de la universidad del país vasco y el actual presidente de la SEP y también del doctor JUAN JOSÉ SANZ tenemos hoy aquí como moderador científico titular en el centro nacional de biotecnología del consejo superior de investigaciones de américa
Nuestros participantes son la doctora LORENA AGUILAR del instituto de investigaciones biomédicas de Mexico la doctora MÓNICA LAMAS del centro de investigación y estudios avanzados también de MEXICO la doctora ALEJANDRA LOYOLA de la fundación ciencia y vida de CHILE y la doctora MARÍA FABIANA DRINK coach de la universidad nacional de Rosario Argentina
Todos han oído hablar del ADN y de nuestros genes.
Esto es un modelo de la doble hélice de ADN transporta la información genética que está contenida en esta molécula y está dentro de todas nuestras células y determina como somos como son nuestras características morfológicas y fisiológicas y muchas veces también qué tipo de enfermedades vamos a padecer.
Si la desarrollamos el ADN vemos que es una secuencia de componentes químicos cuatro diferentes que van formando una lista y cada uno de nuestros genes, nuestras información genética está compuesta por una combinación diferente de todas estas letras
Hasta hace poco pensábamos que ahí estaba todo Pero hay algo más y a esta molécula de ácido nucleico que forman nuestros genes del ADN se pegan proteínas que son las que hacen nuestra. funciones.
Estas moléculas, se pegan y hacen que el ADN transmita esa información a otras moléculas.
De forma que estas proteínas se pegan a la cadena de ADN aquí manejan como funcióna la cadena de ADN y cada una de sus secuencias.
Son como etiquetas de modificaciones químicas.
LA EPIGENÉTICA LO QUE ESTÁ POR ENCIMA DE LA GENÉTICA
La genética es la secuencia de nuestros genes, la epigenética es lo que se pone sobre ellos regulandolos.
El tipo de etiquetas que a veces tiene también el propio ADN va a hacer que se
comporte de una manera o de otra y que algunos genes se activen funcionen y otros genes no funcionen correctamente.
Estas etiquetas se colocan dentro de las moléculas y nos hacen entender cómo funciona la salud.
Muchas otras cosas son vitales para el funcionamiento de la células no solo en humanos, sino también en animales y plantas.
Y asi podemos entender e incluso manipular las características de un ser vivo.
John Gurdon y Kim premios Nobel observaron hace 75 años,
que las distintas células que componen los organismos, tales como neuronas, linfocitos, células hepáticas células tienen la misma información genética el mismo ADN , sin embargo son tremendamente diferentes .
En los 50 se pensaba que la diferenciación celular se debía a que la célula perdía información genética, y entonces una célula muscular iba a tener
información genética distinta a una la neurona porque perdía
información y eso le daba la diferencia, pero en ese año se hizo el primer planeamiento el primer ciclo no una rana desde se sacó desde una célula que ya pertenecía a una rana adulta y se pudo hacer una nueva rana lo que significa la importancia de ese experimento fue que se demostró que esa célula que estaba diferenciada en una rana adulta contenía toda la información de una que podía generar una nueva rana por lo tanto no había pérdida de información entonces. 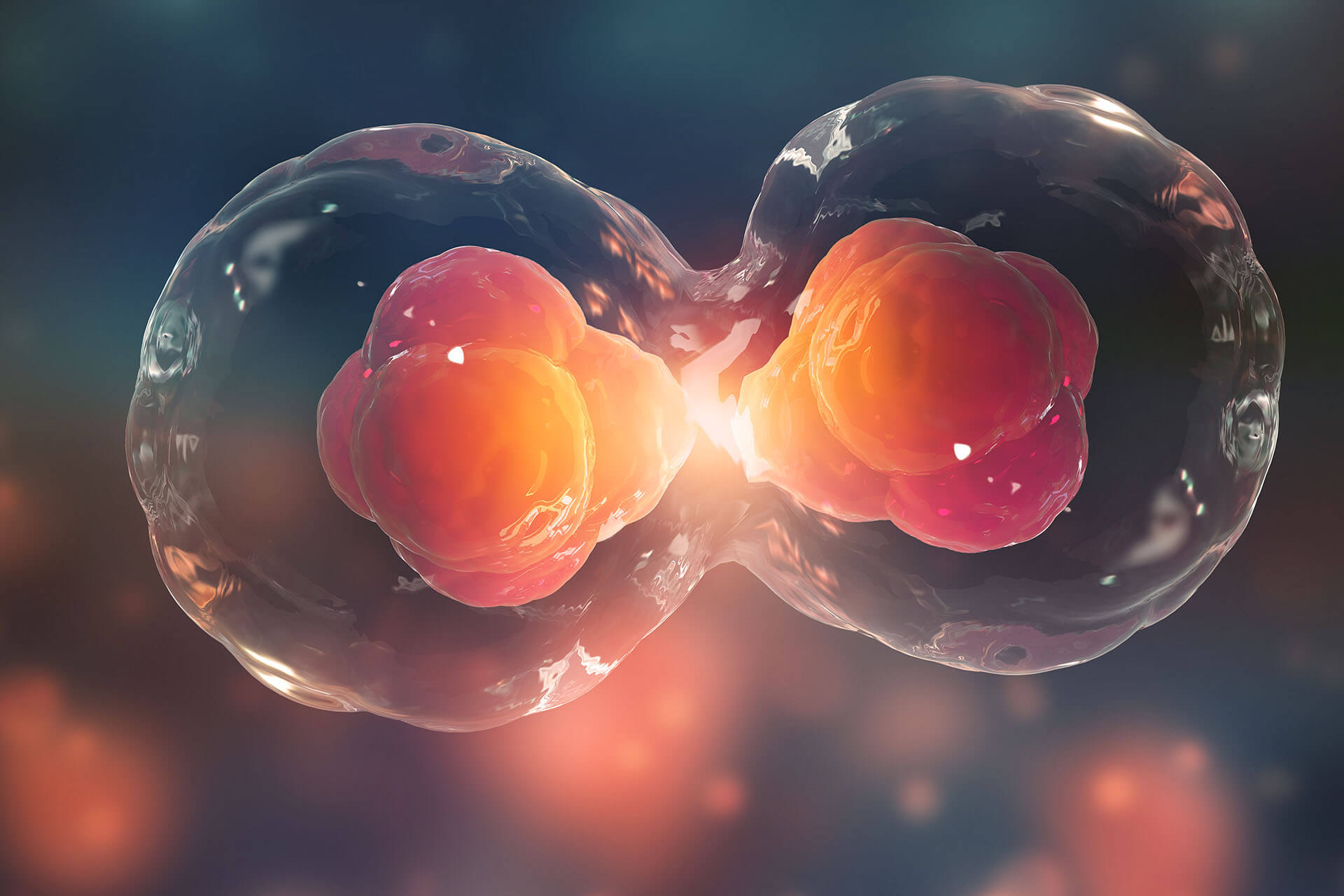
La siguiente idea que una célula neuronal una célula muscular tiene la misma información genética pero expresa distintos genes y eso se regula a nivel genético entonces, ese es un experimento que fue tremendamente importante para definir que hay algo más en el ADN que permite entonces tener distinta información en distintas células.
Estas modificaciones pueden estar tanto en la propia secuencia del ADN como en las proteínas que sintetiza
Estas modificaciones estas marcas que se ponen en el ADN o en las proteínas asociadas lo que hacen es que cambian la arquitectura del DNA vale entonces digamos la imagen muy sencilla de entender
Es que el ADN está formado por ladrillos, cuatro ladrillos que son idénticos y que se repiten en un orden determinado y una serie de grupos químicos que se repiten en un orden determinado uno con unos ladrillos puede hacer a un apartamentito a las afueras de Madrid o el palacio de Linares y esto es lo que hacen estas marcas se fijen en la epigenética se refiere justamente a esto a la arquitectura del DNA cómo está organizado así lo que tienes en una zona determinada es el apartamento a las afueras de Madrid o en el palacio de Linares y tienes una neurona funcionando a todo lo que da entonces.
Durante mucho tiempo los biólogos moleculares con mucho detalle justamente como de mención a Alejandra cómo se expresan los genes como hacer que un gen se produzca cómo se produce la proteína y era como si viéramos durante mucho tiempo una parte muy pequeña de algo y de repente alguien hubiera quitado una cortina y hubiera visto que hay toda una historia detrás inmensa que no considerábamos y que entonces ahora tenemos que considerar porque la imagen es diferente no es lo mismo estar estudiando una parte de un dedo de una estatua y luego quitar la cortina y ver que lo que tienes es el David de Miguel A
Esto es lo que ha hecho la epigenética y estos cambios son los que tenemos que estudiar. sobre lo que estudiábamos antes .
Una nueva visión esta parte del iceberg que está por debajo del agua esto es lo que estamos estudiando y esto es lo que está afectando absolutamente a cómo vemos las enfermedades cómo entendemos el funcionamiento de las células etc porque estos cambios que estamos mencionando que son muy muy variados por una parte es decir hay muchas de estas etiquetas muchas marcas pero además son muy dinámicos.
Una de las características más importantes que realmente realzan la relevancia de lo que es la epigenética es precisamente eso lo que es un mecanismo que tiene gran plasticidad y gran dinamismo y es
reversible porque lo que es el genoma que nos enseñaban y lo que nos enseñaban antes.
Con esta molécula el genoma posibilita que el DNA funcione y se generen los procesos que el propio DNA debe llevar a cabo son estos mecanismos epigenéticos porque ellos son los que tienen movilidad plasticidad
cambian a dependiendo del tipo celular dependiendo de los estímulos los estímulos ambientales son los que permiten que el DNA ejerza su función adecuada en cada momento en cada tipo de celular y en respuesta a cada situación ambiental en el en la cual se encuentra la célula y quizás la característica que yo encuentro más atrayente para para dedicarme precisamente al estudio de la tijera gen ética sí creo que esto que menciona Lorena.
Es muy importante el tema de las
mutaciones y el tema de la plasticidad el epigenética por supuesto que la secuencia del ADN puede cambiar,
puede haber un error, nosotros estamos preparados para que este modelo sea la doble hélice está pensada precisamente
para que se copie a sí misma cada una de las dos cadenas se separan y cada una
sirve de molde para que la otra cadena como si fuera un espejo se copie sobre ella.
Esto es un proceso
físico y puede haber errores y las células están
preparadas para corregir esos errores pero a veces no pueden con ello entonces hay factores que son capaces de
introducir mutaciones en este ADN no ocurren porque
estamos preparados para ello y si ocurren pueden dar lugar a enfermedades.
Hay factores como puede ser por ejemplo la luz ultravioleta del sol
que puede dañar el ADN y por eso tenemos que protegernos porque esas mutaciones se quedan fijadas en el ADN una vez que
ya han sucedido y se transmiten de una célula a la siguiente en cambio todo este dinamismo de la epigenética todo
de las etiquetas moleculares también está muy influido por cosas por factores
ambientales que hasta ahora no éramos muy conscientes y que ahora estamos aprendiendo que no solamente por ejemplo
en este caso pues la luz en sí misma sino que la alimentación la polución la
contaminación hay muchos factores externos que pueden tener un impacto
Hay marcas que dicen que un determinado gen se va a expresar o que un determinado gen no se va a expresar y entonces
nosotros lo que hacemos es estudiar esas marcas para saber este si un gen determinado se va a expresar o no.
En las
plantas por ejemplo se ha visto qué no florecen en el
invierno y se ha visto que está implicada la epigenética en eso se ayuna hay una proteína que se expresa en
determinado momento durante el invierno y hace que no se desencadene la floración y cuando cambia el ambiente
cambia la temperatura cambian determinadas condiciones o las condiciones de luz éste se produce una
marca epigenética que hace que una proteína se exprese y entonces la planta florezca y lo maravilloso que se ha
visto también de estas marcas es que por ejemplo en las plantas se heredan o sea es una memoria que si una determinada
planta por ejemplo se vivió en un determinado ambiente y tiene determinadas condiciones
de generaciones de las plantas esas marcas se van manteniendo o sea no sólo se van manteniendo la secuencia del ADN
sino que por un tiempo hasta que se marque se desmarquen esas marcas también se heredan entonces es esto de un área
que nos ha cambiado un poco este el paradigma de cómo estudiar algunos aspectos cómo abordar algunos
estudios en bioquímica y en biología molecular que nos ha impactado a todos los científicos pero sí que me gustaría
añadir qué esto qué ocurre en las plantas ocurre también en los humanos y qué es una de
las cosas que justamente la epigenética incide hablando de la pregunta que nos
hacemos hoy de cómo incide sobre nuestra salud lo que hacemos nosotros hoy puede afectar a nuestros nietos y por ejemplo
y esto ha habido experimentos hechos en gente que pasó hambre durante la segunda guerra mundial la famosa hambruna en
Holanda hay gente que pasa mucha hambre por el racionamiento de la alimentación los nietos de esta gente hoy en día
podemos seguir las marcas epigenéticas que causó aquella condición y los nietos todavía tienen marcas
epigenéticas en su ADN, o sea que su ADN funciona de una manera particular eso es histórico así fueron los primeros
experimentos que se hicieron sobre estas era de habilidad en el humano y esto implica que hoy en día lo que nosotros
hacemos lo que bebemos lo que comemos el atasco de la m-30 en vivir en la ciudad de México en fin estas cosas
influyen sobre mí epigenoma l y el de nuestros descendientes también y
es una cosa más de las que tenemos que tener en cuenta que antes no
conocíamos esto que mencionas Mónica es muy interesante porque ciertamente hasta
eso significa que modificaciones de nuestro ambiente son capaces de pasar a
generaciones posteriores y eso digamos que era una va en contra del dogma de la
de la herencia genética pero hoy en día efectivamente hay experimentos que demuestran que estas marcas epigenéticas
algunas la mayoría son muy dinámicas son lo que llamamos reversibles son de quita
y pon pero hay otras que son tan estables que incluso pueden pasar las generaciones es cosas y verdad hay experimentos el que has mencionado y
otros en el laboratorio en el que esto se ha demostrado en el en el yo conozco algunos en particular en el área de la
psiquiatría en la que justamente hay una idea de que digamos estas marcas
epigenéticas al principio me digan por ejemplo en el mundo de las adicciones
Las adicciones causan cambios en el epigenoma de la gente, son de estas marcas epigenéticas de la persona adicta
y estas marcas pueden ser reversibles durante un tiempo pero si se mantienen y se mantienen al final se hacen estables
y esas marcas epigenéticas se pueden pasar a la progenie incluso la progenie
la puede pasar a la siguiente progenie efectivamente esto ya está comprobado está comprobado que tenemos un nivel
adicional ya no es sólo para nosotros sino para nuestros hijos nietos etc
Querria que me comentara si alguna en
algún caso concreto de enfermedades por ejemplo en humanos que tengan una base
genética y que esté muy claro muy estudiado alguien tiene a lo mejor incluso trabajáis directamente en vuestros laboratorios en este campo
bueno yo creo que es un campo que el campo de la ficción ética es un campo relativamente nuevo que
no permite o sea es muy complejo porque hay muchos factores que
influencian hay mucha maquinaria molecular implicada muchas proteínas distintas muchas integraciones redes
genéticas que los coordinan y por tanto el entenderlo pues todavía estamos en
ello en lo que son lo que se denominan enfermedades complejas como por ejemplo:
El tema de la obesidad o el desarrollo de diabetes a enfermedades como enfermedades neuropsiquiátricas que tienen muchos factores que convergen para que se dé la enfermedad pues tiene un componente genético muy fuerte pero es de momento nos está resultando muy difícil discernir digamos encontrar el elixir hola e la diana molecular que nos va a permitir resolver estos problemas pero algunos avances se hacen por ejemplo en cáncer algunos tipos de cáncer lo que serían los tratamientos para curarlos pues hay algunos tipos de tratamiento epigenéticos que permiten alguna de la máquina molecular implicada en la en el control del epigenoma y pues han probado efectivos para el tratamiento de ciertos cánceres de ciertos cánceres muy concretos y bueno yo creo que ahí el campo es donde quizás esté más avanzado
La mayoría si no todas las enfermedades finalmente se reducen a que hay un cambio en la expresión de algunos factores y eso está regulado por la parte genética y por la parte de genética
yo diría que los años noventa la investigación estuvo centrada en identificar o sea purificar desde una célula que crece en cultivo llegar hasta la proteína que producía la función
El hilo es buscar qué proteína ponía esta tapita verde acá y entonces fueron muchísimos años décadas donde se hizo eso muchísimos laboratorios la ciencia se hace en conjunto y nosotros tenemos mucho mucha comunicación entre los científicos publicamos nuestros datos entonces ustedes pueden buscarlo es disponible una vez que se identificaron estas proteínas que son tremendamente relevantes
Los científicos nos empezamos a dar cuenta que muchas de esas proteínas estaban montadas en algunas enfermedades la mayoría de las proteínas que van a producir la tapita verde la tapita amarilla roja están mutadas en muchas enfermedades son diferentes o funcionan más o funcionan menos o no funcionan Enfermedades como el cáncer son enfermedades multifactoriales no se debe a una modificación a una a una falla sino que son muchas entonces es difícil definir que una proteína sea responsable hay enfermedades donde si uno puede decir un factor. La hemofilia por ejemplo se debe a que el factor 8 no se produce y eso es porque hay un problema a nivel de ADN puede ser o también puede ser un problema a nivel de la regulación de como ese factor se expresa o es que los mecanismos son tan complejos son multifactoriales son multifactoriales
hay casos de enfermedades monogénicas que es como nos referimos a ellas que un único gen o una única un único problema en un factor concreto causan la enfermedad y puede ser una enfermedad gravísima si lo que está modificado lo que no funciona bien es muy importante pero normalmente las enfermedades sobre todo las enfermedades más prevalentes las que afectan a la mayoría de la población suelen tener origen es multifactorial es entonces muchas veces es muy difícil acotar cuál es el problema y estamos hablando a lo mejor simplemente de la secuencia del ADN si ya metemos todo el tema epigenético es complicado pero es que es justamente ahí, es justamente porque hubo un momento que todos recordarán a veces la ciencia salta del laboratorio a la calle y uno empieza a ver en los periódicos el intermediario que se habla de genes y que hoy en
el gimnasio te hacen el análisis genómico para ver si vas a poder subir tres escaleras o no en fin y hubo un momento en el que todos pensábamos que el DNA era definitivo o sea que tú nacieras con un DNA y esa era tu destino incluso había una película no sé si recuerdan llamaba guataca en o que el de línea fija va a tu destino en la vida y ya no podía ser astronauta si no tenías un cierto DNA y justamente había preguntas en ese
entonces en este concepto por eso se lanzó el proyecto del genoma humano. Vamos a leer el genoma humano entero vamos a ver todos los ladrillos que constituyen el genoma humano y lo vamos a entender y vamos a solucionar todo y vamos a entender todas las enfermedades que no se conocían porque vamos a encontrar todos estos genes que no están funcionando y entonces se resuelve el genoma humano se lee y diez años después seguimos más o menos exactamente en la misma posición no entendemos nada ni sabemos nada de estas enfermedades esto es justamente lo más interesante en los laboratorios cuando un experimento no sale como uno espera, entonces te tiene que poner a pensar y entonces en este caso todas esas preguntas que no se podían resolver porque dos gemelos que tienen exactamente el mismo DNA el mismo DNA no son iguales y uno a los veinte años desarrolla esquizofrenia y el otro no porque no hemos conocido más solución a enfermedades no después de haber leído todo el genoma humano y habernos gastado millones de dólares y porque hemos avanzado muy poco en el conocimiento pues porque las cosas no iban por ahí no rascó y cómo y cómo era esta historia mágica de COMO el ambiente interfiere en el DNA.
Porque un gemelo es esquizofrénico, es el ambiente pero no teníamos ni la más remota idea de cómo el ambiente intervenía
Todo eso es la epigenética como el ambiente interfiere como ya en el genoma
nos dicta nuestro futuro porque el epigenoma como bien dicen todas mis
compañeras se puede cambiar y si tú puedes afectar tu epigenoma y puedes cambiar el destino de esto y en
las enfermedades pues en particular mucho esfuerzo realizado para encontrar
un gen que fuera responsable de una enfermedad estudiando a las familias el afectado la familia a la abuela la
bisabuela que viene de la argentina el otro sector mont y no se encontraban los genes porque porque no estaba en los
genes estaba en la epigenética estaba en la arquitectura estaba en estas marcas que hacen que el genoma sea diferente
La epigenética ha venido a cambiarnos la perspectiva
la epigenética es un campo muy nuevo no entonces pues todavía muchas cosas por
describir y por descubrir y creo que te va a dar muchos más frutos de los que
está dando y todavía hay muchas cosas que podemos entender y que nos van a ayudar a comprender ciertas enfermedades
de relevancia mundial ahora mismo en esa misma línea
Particularmente de los gemelos que es el paradigma de las personas genéticamente idénticas y es así y muchas veces o el
argumento que se ha utilizado históricamente para defender esto es gemelos a lo mejor que son separados y
luego son iguales en aficiones en comportamental pero tú has mencionado el caso contrario que efectivamente es lo que nos estamos dando cuenta casos en
los que son idénticamente genéticos genéticamente igual pero por la epigenética en este caso han cambiado y
en ese sentido creo que también hay estudios que hablan de una genética de la edad es decir la edad el
envejecimiento o cómo vamos avanzando a lo largo de la vida parece ser que hay marcas concretas identificadas que
caracterizan esto creo que eso es así verdad alguien quiere comentar algo
bueno sí o sabe que es así así es claro que todavía no sabemos exactamente
desgraciadamente así es que tenemos marcas verdad pero no sabemos cómo eliminarlas digamos
por lo menos en mamíferos en plantas cómo está el asunto
el ambiente cambió muchísimo el epigenoma las marcas que se ven en distintos ambientes en plantas que
tienen el mismo adn es osea cambia muchísimo de acuerdo a si la
planta crece por ejemplo con más tiempo de luz con menos tiempo de luz se ven marcas distintas y se expresan genes
distintos y el ADN es el mismo siendo a esto que contabas hay un experimento
también clásico con ratones este en los cuales a las madres tienen a los ratones
con el mismo este mismo material genético pero la alimentación de la
madre durante el embarazo influye en que el ratón salga distinto y salga con por ejemplo este exprese una
determinada enfermedad o no y en la alimentación de la madre que le transmite al hijo y salen distintos la
descendencia de acuerdo a ese proceso sea que el ambiente la alimentación donde nos movemos influye muchísimo en
estas marcas influye muchísimo después en lo que se va a expresar.
A
medida que cuando somos embriones y las células empiezan a diferenciarse se van añadiendo distintas marcas para que
una célula sea de una manera o de otra ahora hemos aprendido mecanismos para borrar
esas marcas y volver hacia atrás de forma que una célula adulta sea capaz de
volver hacia un estado pluripotente esto significa que esa
célula ahora la podemos convertir otra vez en otra célula diferente porque
estas marcas que se han ido añadiendo más o menos conseguimos
borrarlas y ahora utilizando un cóctel diferente les convertimos en otra cosa .
El edil es del premio nobel de Yamanaka por este proceso por el que introducía
cuatro genes en una célula y la convertía en una célula primigenia digamos una célula capaz de dar lugar a
todas las células del organismo le dieron el premio nobel y tropecientos mil laboratorios en el mundo
empezaron a tratar de justamente utilizar esta técnica para tener células una fuente ilimitada de
células para poder generar con las células que a mí me hicieran falta que produjeran insulina o células de
neuronas para la enfermedad de Parkinson.
Muchos laboratorios
y de repente se empezaron a dar cuenta de que estas células
si hacían lo que querías que hicieran durante un tiempo y luego dejaban de hacerlo y volvían a su estado
original más o menos y alguien en estado muy poético dijo estas células tienen
memoria epigenética.
El propio Yamanaka después de que le dieron el premio nobel dijo bueno sí claro esto es lo que llevo descubrió muy
importante pero claro tiene también aquellos sus cursos sus problemas
vamos el mensaje es por supuesto el avance es impresionante y es un poco lo
que tú decías al principio se generan nuevas preguntas qué es esta memoria epigenética si yo puedo de verdad
solucionarse a que la célula ya se olvide de lo que fue y que realmente se cometan hacerlo por impotente habrá un
pasado gigantesco o sea que todavía hemos conseguido saber que estas marcas son
importantes y que borrando podemos volver a un estado primigenio pero no es tan sencillo es mucho más complejo.
Al principio los genes cuando se empezó a estudiar el genoma humano nos dimos cuenta de que el porcentaje de toda la
cadena de ADN que tenía información genética transmisible a una proteína es
decir algo que sabíamos que funcionaba era muy pequeña y que la mayoría del ADN
pues no se sabía para qué estaba ahí y dijo no sé nada de basura no sirve para nada este término que es bastante
desafortunado
.
Hoy sabemos que puede ocurrir en zonas del ADN que cuando las leemos pues parece que no son nada y tienen muchas
implicaciones evolutivas porque claro así es como la evolución va trabajando es decir modificando no a lo
mejor la propia información sino la cuándo se tienen que expresar los genes
es lo que lo que hace diferentes unas especies de otras.
Esto
es el aspecto l tridimensional del genoma pues es una nueva capa regulatoria que
se viene estudiando desde hace relativamente poco que también se considera como una capa
epigenética porque no depende de la secuencia del DNA , es cómo se empaqueta el DNA dentro de un núcleo es
decir cómo se dobla como no es lo mismo que bueno sabemos que es una doble hélice que es una doble hélice que se
enrolla sobre sí misma pero luego las estructuras de orden superior es
decir aquellas que tienen un grado de complejidad mayor
Son dos metros de molécula que se empaquetan en
100 nanómetros entonces que es o que tienen el núcleo de diametro
como se dobla esa molécula dentro del núcleo es se está viendo que es algo muy
una pregunta muy relevante y que es realmente
una cuestión que pues determina muchos
aspectos de el desarrollo del embrión o por ejemplo a un tema como la
polidactilia no a la polidactilia se ha visto que a depende hay una zona del
genoma que determina pues de cuántos dedos tenemos en la mano si esa zona del genoma se empaqueta diferente es decir
se dobla sobre sí misma de una forma aberrante se generan fenómenos como la
polidactilia y pues ya se está viendo que hay muchas pues muchas enfermedades
muchos aspectos de la biología que dependen específicamente de cómo se enrolla el dna dentro del núcleo y bueno
pues si es una área de estudio que mi laboratorio pues está explorando no si es un poco
sí porque una de las áreas más activas es estas comparaciones que se hacen entre distintas especies y estas estas
zonas volviendo un poco a las técnicas que utilizamos de una manera muy muy sencilla allí e intentando ya llegar un poco más
a un detalle como digo sin abandonarla a la parte divulgativa conocemos por ejemplo marcas específicas que hacen
cosas concretas es decir sabemos que una proteína de las que se pegan al adn de las que normalmente están pegadas al adn
dándoles parte de esta estructura primaria que yo tiene para que se organice pues hay por ejemplo dos o tres
modificaciones en esa proteína y esas modificaciones más o menos hasta donde sabemos siempre hacen lo mismo ejemplo
activar un gen cuando digo activar es hacer que algo ahora que te produce es y
hay otras que al contrario cuando están esas marcas que a lo mejor es la misma proteína pero es otra marca diferente
como si tuviera dos tapones uno en un sitio y en otro de distintos colores hace pues que no se nos expresen y
estudiando este tipo de marcas comparando unas especies con otras somos capaces de identificar regiones por
ejemplo que son importantes que están conservadas y es una de las cosas con las que se
estudian cuco cómo se como se hace en estos estudios es decir de una manera
sencilla qué tipo de técnicas utilizadas por ejemplo alejandra para saber de detectar por claro nosotros aquí vemos
el tapón de color verde pero como en un laboratorio tú puedes decir pues veo esta marca si antes de hacer un
comentario con respecto con lo que dijiste que es súper relevante nosotros
pensamos que somos especiales pero la verdad es que los mecanismos moleculares que ocurren en nosotros o en una mosca o
en una hormiga son exactamente los mismos el adn
en la bacteria en el mismo los componentes son los mismos lo único que
cambia es la organización entonces hay mucha homología a nivel
molecular en los procesos que ocurren tanto en humanos como en especies como
ratón o qué sé yo mosca y debido a eso podemos usar esos modelos
para investigar hay gente que usa gusanos hay gente que usa levadura hay
gente que usa células humanas y finalmente llegamos a hay variaciones
pero finalmente los mecanismos que ocurren en las distintas especies son los mismos ahora como estudiamos
infinidad de técnicas en mi laboratorio somos bastantes bioquímicos lo que nos gusta es purificar esto o sea a partir
de una gran cantidad de células por ejemplo y llegar a esta proteína y
caracterizar esta proteína investigar cómo funciona en el tubo ensayo si yo le
pongo más temperaturas y le pongo en más azúcar etcétera qué pasa
y una vez que uno entiende esos mecanismos muy básicos puede ir a empezar a variar los a modificar lo
bueno qué pasa si yo le cambio en la la secuencia en desde que tenga este
tubo así que cortar lo que pasa con esa proteína porque eso también nos va a ayudar a entender cómo funciona y quizás
llegar a entender una enfermedad no porque cambia utilizamos entonces métodos de
purificación en este momento debido a toda la tecnología que existe ya hay por
ejemplo secuenciación masiva lo que decías tú recién de la secuenciación del
genoma humano fue además de todo el conocimiento que nos trajo fue una un
abrir y una ayuda técnica pero impresionante y en este momento ya
podemos secuenciar el genoma completo en es barato lo podemos hacer en cualquier
momento podemos secuenciar infinidad de especies en el mismo
momento entonces las técnicas han avanzado muchísimo y ya no estudiamos solamente una
en un momento cuando yo partí mi estudio investigamos esta región pero en este
momento ya se investiga el genoma completo de las células y al mismo tiempo se estudia estas marcas a nivel
de todas las células ya no solamente en una región chiquitita sino que a nivel
general y eso también a seguir es favorecido por técnicas que parten
como investigación básica y después a una investigación a un investigador se le ocurre que se puede aplicar en otra
cosa y entonces así se van generando conocimientos nuevo y también técnicas nuevas que ayudan muchísimo no puedo
entrar en detalle porque no tienen ningún sentido general
minutillos su minuciosa y extraordinaria los que somos un poco más perezosos hacemos otras cosas
utilizamos técnicas que te permiten ver estas marcas sin necesidad de tener que purificarlo porque de verdad es un
trabajo extraordinario de tener que purifica las cosas entonces puedes utilizar una proteína que reconoce la
tapita verde y esa le pega es una cosa que es fluorescente y entonces puedes verlo en la célula en vivo en el
microscopio es la marquita cómo se mueve dentro del núcleo eso hacemos los que hacemos un poco más precios no hay que
aleja nada son técnicas complementarias son y también y en cuanto a la comparación con las especies por ejemplo
nosotros vamos a utilizado en el laboratorio para un problema de regeneración por ejemplo los peces
regeneran la retina y los mamíferos no entonces nosotros queríamos saber qué es
lo que tiene el pez que no tenemos nosotros o qué es lo que tenemos nosotros que molesta y que no debería estar en cuanto a epigenética porque
teníamos pistas de que esto podría ser un problema genético y lo que hicimos fue comparar directamente lo que es lo
que hacen las cosas epigenéticas que ocurren algunas que ocurren en el pp después del daño para que mientras va
regenerando la retina y lo que hace el ratón por ejemplo el mamífero y
buscábamos obviamente diferencias y no similitudes y encontramos algunas cosas que ocurren igual y otras que son
totalmente diferentes cientos nos vamos a poner esas claro en esas actividades epigenéticas que están haciendo si yo la
puedo modular en el laboratorio a lo mejor puede hacer que el ratón regenere la retina
y bueno obviamente hay muchas utilizas el conocimiento decía alejandro muy bien
que la ciencia la hacemos todos en conjunto con muchos laboratorios trabajando utilizas la información que
publica otra gente otros laboratorios para crecer tu propia investigación y
hacer una escalera que va conduciendo no sé si en este sentido otra cosa importante que también hemos oído y
muchas veces le ponemos apellidos a la ciencia ciencia básica ciencia aplicada y a veces por supuesto que queremos
solucionar los problemas y queremos hacerlo lo antes posible pero quizás sea equivocado centrarnos en enfocar la
ciencia en solucionar un problema la ciencia es conocimiento y el conocimiento siempre es importante y siempre va a dar un fruto mañana o
dentro de 20 años en este sentido por ejemplo vuestras investigaciones que pueden parecer que son muy básicas o que
no llevan a ningún sitio o la pregunta que a todos los científicos nos suelen hacer y eso que tú haces para qué sirve sirve para aprender y aprender siempre
está bien y por supuesto aprender cosas de detalle de los que estamos hablando pues pues mucho más querías fabiana
mencionar porque el tema de los anticuerpos a mí me parece importante porque a lo mejor mucha gente ha oído
hablar de anticuerpos como algo de nuestra defensa pero es una de las herramientas más fundamentales que usamos todos los científicos en el
laboratorio a estudiar las marcas epigenéticas por ejemplo si hay una marca amarilla
indicar que un gen se va a activar hay anticuerpos específicos para la marca amarilla la marca roja la marca verde
entonces se se incuban se ponen en distintos ambientes y
después se incuban con esos anticuerpos se precipita se purifica y se identifica que gen está o qué sector del adn está
marcado con esa marca que vamos a saber que se va a activar o se va a inhibir entonces este identificando esas marcas
con anticuerpos específicos de las marcas identificamos qué marcas hay identificamos que hay distintas marcas
de acuerdo en qué en qué ambiente esté esa célula si el tema de los anticuerpos como herramientas de investigaciones es
fascinante y es un ejemplo más de cómo algo que la naturaleza digamos ha producido para un fin que es defendernos
de las infecciones porque recuerdan que un anticuerpo es una molécula es una
molécula que reconoce a otra básicamente entonces normalmente lo que lo que hacen es reconocer por ejemplo
bacterias virus que nos infectan para eliminarlos esa es su función natural si nosotros ahora desarrollamos anticuerpos
contra el tapón amarillo pues somos capaces de las células en el tubo de ensayo usar
estos anticuerpos como una herramienta que nos permite saber y identificar dónde están estos anticuerpos otro tema
que está muy dado muy de moda ahora a lo mejor han oído es el tema de la edición genómica el cris per es una técnica que
lo mismo es una tecnología un ejemplo que unas bacterias desarrollaron para defenderse y que nosotros ahora hemos
utilizado para precisamente cambiar la secuencia del adn de una manera muy
mucho más sencilla de lo que de lo que ocurría hasta hasta ahora
a mí esto me parece tremendamente relevante porque de nuevo con la idea de
que nosotros tenemos que hacer investigación que sea aplicada o que tenga la investigación no necesariamente
nos va a llevar a eso o sea puede que estemos pensando de una manera que no nos va a llevar a ninguna a ninguna parte sin embargo investigaciones como
lo que están mencionando con jose de crisis percas que en la edición higiénica que se está haciendo ahora fueron investigadoras mujeres que
estaban interesadas en invertir en entender cómo la bacteria se defiende o
sea si yo hubiese dicho eso no sé 20 años atrás quizá había medir tubería ti
qué te importa o sea para que no pero sin embargo ese conocimiento está
y bueno después alguien se le ocurrió a espero esto lo podemos utilizar y yo
creo que va a ser una de las revoluciones medicina porque es probable que podamos después editar nuestro genoma no ahora en un tiempo más pero es
una cosa que lo más probable va a pasar y va a revolucionar la medicina entonces uno no sabe por donde la aplicación del
conocimiento va a ocurrir y entonces enfocar la investigación en solamente
aplicación muchos gobiernos lamentablemente en mi país ocurre muchos políticos quieren ver efectos o
resultados inmediatos y la ciencia no es así no es así muy lejos puede que hay un
descubrimiento que se hizo veinte años atrás y que la persona que lo hizo nunca se le ocurrió pero a la persona que lo
leyó a mira esto cómo publicar estas cosas y ahí se genera la aplicación pero
uno no hace una investigación basada en aplicación yo creo que eso es un error que mucha gente tiene y por ahí no va o
sea no podemos no existe la ciencia aplicada no existe es la generación de
conocimiento y en algún momento eso va a llevar aplicación es algo que uno tiene que entender y que
darle los tiempos a los científicos para que investiguen no son los mismos tiempos los políticos que quieren
reelección y entonces quieren resultados de ciencia tiene otros tiempos que son
hermosos no porque nos da tiempo para investigar pero pero no es la
investigación es generación de conocimiento con la potencialidad de
utilizarla
ue está dentro de todas nuestras células es lo que determina pues bueno como somos como son nuestras
características morfológicas cómo son nuestras características fisiológicas muchas veces también qué tipo de
enfermedades vamos a padecer hasta hace poco pensábamos que aquí estaba todo aquí es cuando digo aquí en esta
molécula que normalmente tiene una estructura enrollada como una doble hélice pero si la desarrollamos vemos
que es una secuencia si ven a ustedes aquí una secuencia de componentes
químicos cuatro diferentes que van formando una lista y cada uno de nuestros genes cada uno de
nuestras información genética está compuesta por una combinación diferente de todas estas letras para hacer un
paralelismo aquí están dibujado con colores pues el hecho de que una persona tenga aquí amarillo azul amarillo rojo
bueno pues esa secuencia que son nuestros genes determina como somos y como digo hasta hace poco pensábamos que
ahí estaba todo ahora hemos descubierto que hay algo más y hay algo más
significa que a esta molécula de de ácido nucleico que forman nuestros genes del adn se pegan proteínas son otras
moléculas que son las que hacen las funciones de nuestra en nuestras células y estas moléculas estas proteínas se
pegan y hacen que el adn transmita esa información la información que está aquí contenida está secuencia tiene que
transmitirse a otras moléculas y estas proteínas que se pegan aquí manejan como
función como como es como ocurre esto pues entonces estas proteínas a su vez hay muchas de ellas y pueden tener una
serie de etiquetas por ejemplo aquí hay una que tiene una etiqueta verde una etiqueta amarilla y una
que te arroja para simplificar hablo en colores ustedes imaginen que lo que estamos hablando aquí son de modificaciones químicas bueno pues esto
es la epigenética epigenética lo que está por encima de la genética la genética es la secuencia de nuestros
genes la epigenética es lo que se pone sobre ellos regulando los y de esto es lo que vamos a hablar hoy porque qué
tipo de etiquetas tienen estas proteínas qué tipo de etiquetas a veces tiene también el propio adn va a hacer que se
comporte de una manera de otra y que algunos genes se activen funcionen otros
genes no funcionen correctamente y sabiendo cómo se controla cómo estas
etiquetas se colocan dentro de las moléculas podemos entender pues muchas cosas de cómo funciona la salud porque
quizás es lo que más nos importa pero muchas otras cosas por ejemplo también las plantas los animales tienen
epigenética y controlando estos mecanismos pues podemos entender e
incluso llegar a manipular en nuestro beneficio las características que tienen
entonces hecha esta breve introducción para ponernos en contexto voy a preguntarles a nuestras invitadas
es como se descubrió cuando se empezó a
entender que este tipo de modificaciones existían y de qué manera podemos
estudiarlas alejandra por ejemplo cuando surgió digamos este conocimiento cuando nos dan
nos dimos cuenta de que había estas marcas moleculares yo creo que un hito
importante en este área aunque todavía no se mencionaba como ty genética fueron unos estudios en los años 50 donde por
un premio nobel actualmente john gurdon y kim que ellos estaba la idea de que
las células tenemos distintas células nuestro cuerpo tenemos neuronas tenemos linfocitos que
son los que nos ayudan a responder frente infecciones tenemos células hepáticas que están en el hígado
todas estas células tienen la misma información genética el mismo adn
sin embargo son tremendamente diferentes y en los 50 se pensaba que la
diferenciación celular o sea que nosotros tengamos estas distintas células se debía a que la célula perdía
información genética entonces una célula muscular iba a tener
información genética distinta a una célula de en la neurona porque perdía
información y eso le daba la diferencia pero en ese año entonces se hizo el
primer planeamiento el primer ciclo no una rana desde se sacó desde desde una
célula que ya pertenecía a una rana adulta y se pudo hacer una nueva rana lo
que significa la importancia de ese experimento fue que se demostró que esa célula
que estaba diferenciada en una rana adulta contenía toda la información de
una que podía generar una nueva rana por lo tanto no había pérdida de información entonces cuál es la siguiente idea que
una célula neuronal una célula muscular tiene la
misma información genética pero expresa distintos genes y eso se regula a nivel
genético entonces para mí ese es un experimento que fue tremendamente importante para definir que hay algo más
en el adn que nos permite entonces tener distinto información en distintas células
he mencionado brevemente no sé si alguna puede contribuir a esto que realmente estas modificaciones pueden estar tanto
en la propia secuencia del adn como en las proteínas mónica por ejemplo puedes comentar un
poquito bueno yo quisiera añadir un poco a esta a esta imagen que tú nos has dado
que estas modificaciones estas marcas que se ponen en el adn o en las
proteínas asociadas lo que hacen es que cambian la arquitectura del dna vale
entonces digamos la imagen muy sencilla de entender es que el dna está formado
por ladrillos cuatro ladrillos que son idénticos y que se repiten en un orden determinado
y una serie de grupos químicos que se repiten en un orden determinado uno con unos ladrillos puede hacer a un
apartamentito a las afueras de madrid o el palacio de linares estamos de acuerdo y esto es lo
que hacen estas marcas estas marcas se fijen en la epigenética se refiere justamente a esto a la arquitectura del
dna cómo está organizado así lo que tienes en una zona determinada es el apartamento a las afueras de madrid o es
el palacio de linares y tienes una neurona funcionando a todo lo que da entonces
esto es importante porque durante mucho tiempo nosotros estuvimos
estudiando nosotros me refiero a los biólogos moleculares con mucho detalle
justamente como de mención a alejandra cómo se expresan los genes como hace que hacer que un gen se produzca cómo se
produce la proteína y era como si viéramos está estudiando durante mucho tiempo una parte muy pequeña de algo y
de repente alguien hubiera quitado una cortina y hubiera visto que hay toda una historia detrás inmensa que no
considerábamos y que entonces ahora tenemos que considerar porque la imagen es diferente no es lo mismo estar
estudiando una parte de un dedo de una estatua y luego quitar la cortina y ver que lo que tienes es el david de miguel
ángel por ejemplo esto es lo que ha hecho la epigenética entonces estos son estos son los cambios que
que nosotros tenemos que estudiar ahora sobre todo lo que estudiábamos antes toda esta nueva visión esta parte del
iceberg que está por debajo del agua esto es lo que estamos estudiando y esto es lo que está afectando absolutamente a
cómo vemos las enfermedades cómo entendemos el funcionamiento de las células etc
porque lorena por ejemplo estos cambios que estamos mencionando que son muy muy
variados por una parte es decir hay muchas de estas etiquetas muchas marcas pero además son muy dinámicos es cierto
es decir si ciertamente yo creo que una de las características más importantes
que realmente realzan la relevancia de lo que es la epigenética es precisamente
eso lo que es un mecanismo que tiene gran plasticidad y gran dinamismo y es
reversible porque lo que es el genoma que nos enseñaban nos enseñaba antes con jose con esta molécula el genoma es el
mismo todo el tiempo en todas las células entonces es una molécula que o sea en sí la composición no varía no
acerca de una mutación en teoría no debería de ocurrir aunque ocurre pero no es algo que sea adecuado
lo que posibilita que el dna funcione y se generen los procesos que el propio de
neal debe llevar a cabo son estos mecanismos epigenéticos porque ellos son los que tienen movilidad plasticidad
cambian a dependiendo del tipo celular dependiendo de los estímulos los
estímulos ambientales son los que permiten que el dna ejerza
su función adecuada en cada momento en cada tipo de celular y en respuesta a cada situación ambiental en el en la
cual se encuentra la célula y bueno pues yo creo que sí que este es quizás la
característica que yo encuentro más atrayente para para dedicarme precisamente al estudio de la tijera
ética sí creo que esto que menciona a lorena es es muy importante el tema de las
mutaciones y el tema de la plasticidad el epigenética por supuesto que la secuencia del adn puede puede cambiar
puede haber un error nosotros estamos preparados para que este modelo sea la doble hélice está pensada precisamente
para que se copie a sí misma cada una de las dos cadenas se separan y cada una
sirve de molde para que la otra cadena como si fuera un espejo se se copie sobre ella claro eso es un proceso
mecánico bueno mecánico físico y puede haber errores las células están
preparadas para corregir esos errores pero a veces no pueden con ello entonces hay factores que son capaces de
introducir mutaciones en este adn en la secuencia inicial como comentaba lorena pero normalmente no ocurren porque
estamos preparados para ello y si ocurren pues pueden dar lugar a enfermedades hay factores como puede ser por ejemplo la luz ultravioleta del sol
que puede dañar el adn y por eso tenemos que protegernos porque esas mutaciones se quedan fijadas en el adn una vez que
ya han sucedido y se transmiten de una célula a la siguiente en cambio todo este dinamismo de la epigenética todo
de las etiquetas moleculares también está muy influido por cosas por factores
ambientales que hasta ahora no éramos muy conscientes y que ahora estamos aprendiendo que no solamente por ejemplo
en este caso pues la luz en sí misma sino que la alimentación la polución la
contaminación hay muchos factores externos que pueden tener un impacto y esto creo que es una de las cosas importantes que hemos aprendido verdad
en fabiana sí porque en estas marcas que determinan si un determinado gen se va a expresar o
no este influye el ambiente el ambiente hace cambia estas marcas y entonces hay
marcas que dicen que un determinado gen se va a expresar o que un determinado gen no se va a expresar y entonces
nosotros lo que hacemos es estudiar esas marcas para saber este si si un gen determinado se va a expresar o no en las
plantas por ejemplo que es un área donde yo estudio se ha visto por ejemplo de por qué las plantas no florecen en el
invierno y se ha visto que está implicada la epigenética en eso se ayuna hay una proteína que se expresa en
determinado momento durante el invierno y hace que no se desencadene la floración y cuando cambia el ambiente
cambia la temperatura cambian determinadas condiciones o las condiciones de luz éste se produce una
marca epigenética que hace que una proteína se exprese y entonces la planta florezca y lo maravilloso que se ha
visto también de estas marcas es que por ejemplo en las plantas se heredan o sea es una memoria que si una determinada
planta por ejemplo se vivió en un determinado ambiente y tiene determinadas condiciones
de generaciones de las plantas esas marcas se van manteniendo o sea no sólo se van manteniendo la secuencia del adn
sino que por un tiempo hasta que se marque se desmarquen esas marcas también se heredan entonces es esto de un área
que nos ha cambiado un poco este el paradigma de cómo estudiar algunas algunos aspectos cómo abordar algunos
estudios en bioquímica y en biología molecular que nos ha impactado a todos los científicos pero sí que me gustaría
añadir qué esto qué ocurre en las plantas ocurre también en los humanos y qué es una de
las cosas que justamente la epigenética incide hablando de la pregunta que nos
hacemos hoy de cómo incide sobre nuestra salud lo que hacemos nosotros hoy puede afectar a nuestros nietos y por ejemplo
y esto ha habido experimentos hechos en gente que pasó hambre durante la segunda guerra mundial la famosa hambruna en
holanda hay gente que pasa mucha hambre por el racionamiento de la alimentación los nietos de esta gente hoy en día
podemos seguir las marcas epigenéticas que causó aquella aquella condición y los nietos todavía tienen tienen marcas
epigenéticas en su adn o sea que su adn funciona de una manera particular eso es histórico así fueron los primeros
experimentos que se hicieron sobre estas era de habilidad en el humano y esto implica que hoy en día lo que nosotros
hacemos lo que bebemos lo que comemos el atasco de la m-30 en vivir en la ciudad de méxico en fin estas cosas
influyen sobre mí epigenoma el pez ya no ma de nuestros descendientes también y
es una una cosa más de las que tenemos que tener en cuenta que antes no
conocíamos esto que mencionas mónica es muy interesante porque ciertamente hasta hasta hace poco y yo creo que hoy en día
y controversia también porque claro eso significa que modificaciones de nuestro ambiente son capaces de pasar a
generaciones posteriores y eso digamos que era una va en contra del dogma de la
de la herencia genética pero hoy en día efectivamente hay experimentos que demuestran que estas marcas epigenéticas
algunas la mayoría son muy dinámicas son lo que llamamos reversibles son de quita
y pon pero hay otras que son tan estables que incluso pueden pasar las generaciones es cosas y verdad hay experimentos el que has mencionado y
otros en el laboratorio en el que esto se ha demostrado en el en el yo conozco algunos en particular en el área de la
psiquiatría en la que justamente hay una idea de que digamos estas marcas
epigenéticas al principio me digan por ejemplo en el mundo de las adicciones
las adicciones causan cambios en el epigenoma de la gente son de estas marcas epigenéticas de la persona adicta
y estas marcas pueden ser reversibles durante un tiempo pero si se mantienen y se mantienen al final se hacen estables
y esas marcas epigenéticas se pueden pasar a la progenie incluso la progenie
la puede pasar a la siguiente progenie efectivamente esto ya está comprobado está comprobado que tenemos un nivel
adicional ya no es sólo para nosotros sino para nuestros hijos nietos etc quería que me comentara si alguna en
algún caso concreto de enfermedades por ejemplo en humanos que tengan una base
genética y que esté muy muy claro muy estudiado alguien tiene a lo mejor incluso trabajáis directamente en vuestros laboratorios en este campo
bueno yo creo que es un campo que el campo de la ficción ética es un campo relativamente nuevo que
pues no permite o sea es muy complejo porque hay muchos factores que
influencian hay muchas mucha maquinaria molecular implicada muchas proteínas distintas muchas integraciones redes
genéticas que los coordinan y por tanto el entenderlo pues todavía estamos en en
ello en lo que son lo que se denominan enfermedades complejas como por ejemplo
el tema de la obesidad o el desarrollo de diabetes a enfermedades como
enfermedades neuropsiquiátricas que tienen muchos factores que convergen
para que se dé la enfermedad pues tiene un componente genético muy fuerte pero
es de momento nos está resultando muy difícil discernir digamos encontrar el
elixir hola e la diana molecular que nos va a permitir
resolver estos problemas pero algunos avances se hacen por ejemplo en cáncer algunos tipos de cáncer
lo que serían los tratamientos para curarlos pues hay algunos tipos de tratamiento tratamientos epigenéticos
que permiten son químicos que modulan
algunas de las de alguna de la máquina molecular implicada en la
en el control del epigenoma y pues han probado efectivos para el tratamiento de
ciertos cánceres de ciertos cánceres muy concretos y bueno yo creo que ahí el campo es donde quizás esté más avanzado
en mi opinión no sé si ustedes conozcan bien yo creo que lo que pasa es que en
la mayoría si no todas las enfermedades finalmente se reducen a que hay un
cambio en en la expresión de algunos factores
y eso está regulado por la parte genética y por la parte de genética
yo diría que los años noventas la investigación estuvo centrada en
identificar osea purificar desde desde una célula que se crece en cultivo
llegar hasta la proteína que producía la función y el hilo como dice juan jose es
buscar qué proteína ponía esta tapita verde acá
y entonces fueron muchísimos años décadas donde se hizo eso muchísimos
laboratorios la ciencia se hace en conjunto y nosotros tenemos mucho mucha comunicación entre los científicos
publicamos nuestros datos entonces ustedes pueden buscarlo es disponible
una vez que se identificaron estas proteínas que son tremendamente relevantes
los científicos nos empezamos a dar cuenta que muchas de esas proteínas estaban montadas en algunas enfermedades
la mayoría de las proteínas que van a producir la tapita verde la tapita amarilla roja están mutadas en muchas
enfermedades son diferentes o funcionan más o funcionan menos o no funcionan
enfermedades como el cáncer son enfermedades multifactoriales no se debe
a una modificación a una a una falla sino que son muchas entonces es difícil
definir que una proteína sea ay hay enfermedades donde si uno puede decir un
factor hemofilia por ejemplo se debe a que el factor 8 no se produce y eso es
porque hay un problema a nivel de adn puede ser o también puede ser un problema a nivel de la regulación de
como ese factor se expresa
y la pregunta se me falta algún ejemplo
concreto y lo que está quedando claro es que los mecanismos son tan complejos son multifactoriales son multifactoriales
hay casos de enfermedades monogénicas que es como nos referimos a ellas que un único gen o una única un único problema
en un factor concreto causan la enfermedad y puede ser una enfermedad gravísima si lo que está modificado lo
que no funciona bien es muy importante pero normalmente las enfermedades sobre todo las enfermedades más prevalentes
las que afectan a la mayoría de la población suelen tener origen es multifactorial es entonces muchas veces
es muy difícil acotar cuál es el problema y estamos hablando a lo mejor simplemente de la secuencia del adn si
ya metemos todo el tema epigenético es complicado pero es que es justamente ahí
es justamente porque hubo un momento que todos
recordarán a veces la ciencia salta del laboratorio a la calle y uno empieza a
ver en los periódicos el intermediario que se habla de genes y que hoy y que en
el gimnasio te hacen el análisis de análisis genómico para ver si vas a poder subir tres escaleras o no en fin y
hubo un momento en el que todos pensábamos que el dna era definitivo o
sea que tú nacieras con un dna y esa era tu destino incluso había una película no sé si recuerdan llamaba gattaca en o que
el de línea fija va a tu destino en la vida y ya no podía ser astronauta si no
tenías un cierto dna y justamente había preguntas en ese
entonces en este concepto por eso se lanzó el proyecto del genoma humano vamos a leer el genoma humano entero vamos a ver todos los ladrillos que
constituyen el genoma humano y lo vamos a entender y vamos a solucionar todo y vamos a entender todas las enfermedades
que no se conocían porque vamos a encontrar todos estos genes que no están funcionando y entonces
se resuelve el genoma humano se lee y diez años después seguimos más o menos exactamente en la misma posición no
entendemos nada ni sabemos nada de estas enfermedades esto es justamente lo más interesante en los laboratorios cuando
un experimento nos sale como uno espera porque entonces te tiene que poner a pensar y entonces en este caso todas
esas preguntas que no se podían resolver porque dos gemelos que tienen exactamente el mismo dna el mismo dna no
son iguales y uno a los veinte años desarrolla esquizofrenia y el otro no porque no hemos conocido más solución a
enfermedades no después de haber leído todo el genoma humano y habernos gastado millones de dólares y porque hemos
avanzado muy poco en el conocimiento pues porque las cosas no iban por ahí no rascó y cómo y cómo era esta historia
mágica de no el ambiente interfiere en el dna porque un gemelo es esquizofrénico nungwi es el ambiente no
pero no teníamos ni la más remota idea de cómo el ambiente intervenía bueno todo eso es la epigenética como el ambiente interfiere como ya en el genoma
no dicta nuestro futuro porque el epigenoma como bien dicen todas mis
compañeras se puede cambiar y si tú puedes afectar tu epigenoma y puedes cambiar el destino de esto y en
las enfermedades pues en particular mucho esfuerzo realizado para encontrar
un gen que fuera responsable de una enfermedad estudiando a las familias el afectado la familia a la abuela la
bisabuela el que viene de la argentina el otro sector mont y no se encontraban los genes porque porque no estaba en los
genes estaba en la epigenética estaba en la arquitectura estaba en estas marcas que hacen que el genoma sea diferente
entonces ahí es justo todo esto es justo donde en la epigenética ha venido a cambiarnos la perspectiva si yo también
quería añadir que en ese misma en esa misma línea que o sea alejándola nos ha
dicho hace un rato que en los años 50 mil o 150 si se comenzaba a hablar de este tipo de conceptos epigenéticas fue
acuñada pues la palabra en concreto por aquella época pero es un campo relativamente nuevo no porque no ha sido
hasta finales de los 90 años 2000 o así que ha empezado a crecer exponencialmente gracias a las nuevas
tecnologías y a las nuevas capacidades que tenemos para entender este tipo de problemas y entonces bueno pues no ha
pasado tanto en un tiempo largo que tiene de historia
la ciencia pues el campo de la epigenética es un campo muy nuevo no entonces pues todavía muchas cosas por
describir y por descubrir y creo que te va a dar muchos más frutos de los que
está dando y todavía hay muchas cosas que que podemos entender y que nos van a ayudar a comprender ciertas enfermedades
de relevancia mundial ahora mismo en esa misma línea
si hay varias cosas mónica que has mencionado que son muy interesantes tenía aquí y de hecho ha notado a sacarle el tema por ejemplo
particularmente de los gemelos que es el paradigma de las personas genéticamente idénticas y es así y muchas veces o el
argumento que se ha utilizado históricamente para defender esto es gemelos a lo mejor que son separados y
luego son iguales en aficiones en comportamental pero tú has mencionado el caso contrario que efectivamente es lo que nos estamos dando cuenta casos en
los que son idénticamente genéticos genéticamente igual pero por la epigenética en este caso han cambiado y
en ese sentido creo que también hay estudios que hablan de una genética de la edad es decir la edad el
envejecimiento o cómo vamos avanzando a lo largo de la vida parece ser que hay marcas concretas identificadas que
caracterizan esto creo que eso es así verdad alguien quiere comentar algo
bueno sí o sabe que es así así es claro que todavía no sabemos exactamente
desgraciadamente así es que tenemos marcas verdad pero no sabemos cómo eliminarlas digamos
por lo menos en mamíferos en plantas cómo está el asunto
el ambiente cambió muchísimo el epigenoma las marcas que se ven en distintos ambientes en plantas que
tienen el mismo adn es osea cambia muchísimo de acuerdo a si la
planta crece por ejemplo con más tiempo de luz con menos tiempo de luz se ven marcas distintas y se expresan genes
distintos y el adn es el mismo siendo a esto que contabas hay un experimento
también clásico con ratones este en los cuales a las madres tienen a los ratones
con el mismo este mismo material genético pero la alimentación de la
madre durante el embarazo influye en que el ratón salga distinto y salga con por ejemplo este exprese una
determinada enfermedad o no y en la alimentación de la madre que le transmite al hijo y salen distintos la
descendencia de acuerdo a ese proceso sea que el ambiente la alimentación donde nos movemos influye muchísimo en
estas marcas influye muchísimo después en lo que se va a expresar quisiera sobre eso es que es muy bonito a la
nutrición y la epigenética porque además se lo van a encontrar en el supermercado cualquiera
las revisiones científicas lo que hacen es tabular poner en una tabla que
componente epigenético se ha descrito por ejemplo que se yo en el caldo de pollo en el ajo el nose que edita yo
cuando hago todas mis clases le digo a estudiantes que toda esta epigenética lo que está haciendo es confirmar que la
abuela tenía razón y que la isla de bueno comer el brócoli y que no es muy
bueno tomar mucho azúcar y todo esto explicado desde un punto de vista molecular es parte de lo que ha pasado en el congreso esta bioquímica en la
ciudad que han hablado de etapas y la biología molecular de las tapas y la bioquímica de las tapas bueno pues esto
es exactamente iba con la epigenética y cuando digo que se lo encuentra en el que se va encontrar en la calle es real
porque ya ahí si ustedes buscan en internet epigenética y comida por
ejemplo yo busqué en internet en algún momento para dar una clase y había comida para perros epigenética
epigenética no sé que se llama creo que te dan un perro buenísimo y creo que no
hay todavía para humanos y yo le dije a los estudiantes el primero que me traigo un yogurt epigenético le regaló una botella de tequila ya son ya son mayores
mis estudiantes es donde doctorado todos los de aquí y como locos no encontraron porque es mucho más difícil hacer una
regulación de comida para humanos pero de perro ya la pueden encontrar esto es como la ciencia sal salta del
laboratorio a la calle yo creo que por eso estas sesiones son buenísimas porque de esta manera podemos
conocer y saber que nos está tomando el pelo en la opinión crítica claro que si mónica el comida epigenética mejor no
pero jabón epigenético pues con sabor ya pues hay que empezar a sacar la botella
o sea que entonces claro tenemos tenemos unas marcas epigenéticas de envejecimiento que hemos dicho que bueno
es una cosa que nos gustaría a todos aprender más y controlar verdad pero precisamente también por la epigenética
otro descubrimiento fundamental y aquí es muy relevante lo que contaba alejandra al principio sobre el premio
nobel de de jong cartoon porque compartió el premio nobel con otro investigador en este caso japonés shinya
yamanaka que lo que hizo fue algo no es una palabra que un científico debiera
utilizar pero para que me entiendan coloquialmente algo milagroso y es el tema todo el tema de las células madre
inducidas pluripotentes inducidas no sé si se habrán oído hablar de ello y
básicamente lo que lo que consiguió y lo que se ha conseguido y es uno de los campos más activos hoy en día de trabajo
en los laboratorios es precisamente volver atrás este reloj o estas marcas epigenéticas como ha comentado alejandra
a medida que cuando somos embriones y las células empiezan a diferenciarse se van añadiendo distintas marcas para que
una célula sea de una manera o de otra ahora hemos aprendido métodos mecanismos para borrar
esas marcas y volver hacia atrás de forma que una célula adulta sea capaz de
tener volver hacia un estado un estado pluripotente esto significa que esa
célula ahora la podemos convertir otra vez en otra célula diferente porque
porque estas marcas que se han ido añadiendo más o menos conseguimos
borrarlas y ahora utilizando un cóctel diferente les convertimos en otra cosa
veo que mónica no está muy bien es que es justamente mi tema de trabajo vengan las células basales l aún es diferente a
una experta no fue muy importante el edil es del premio nobel de yamanaka por este proceso por el que introducía
cuatro genes en una célula y la convertía en una célula primigenia digamos una célula capaz de dar lugar a
todas las células del organismo me dieron el premio nobel y tropecientosmil laboratorios en el mundo
empezaron a tratar de justamente utilizar esta técnica para tener células una fuente ilimitada ilimitada de
células para poder generar con las células que a mí me hicieran falta que produjeran insulina o células de
neuronas para la enfermedad de parkinson que se yo muchos laboratorios
y de repente se empezaron a dar cuenta de que estas células sí o sea en algunos
casos si hacían lo que tuvo querías que hicieran durante un tiempo y luego dejaban de hacerlo y volvían a su estado
original más o menos y alguien en estado muy poético dijo estas células tienen
memoria memoria epigenética es decir que tú tienes una célula que
tiene una arquitectura tiene una célula que tiene el palacio de linares dentro y tú puedes al revés tienes tu célula con
tu pisito en chamartín y entonces tratas de cambiarlo y tratas de hacer para orinar
y la célula empieza a hacer las cosas empiezan a cambiar empieza a ahorrar marcas empiezan de marcas nuevas pero
tiene la tendencia a volver a ser tu pisito no voy a volver a hacer lo que era eso es lo que se llama memoria
epigenética es que justamente les voy a hablar mañana en el congreso eso entonces muchos investigadores y el
propio yamanaka después de que le dieron el premio nobel dijo bueno sí sí claro esto es lo que llevo descubrió muy
importante pero claro tiene también aquellos sus cursos sus problemas
vamos el mensaje es por supuesto el avance es impresionante y es un poco lo
que tú decías al principio se generan nuevas preguntas qué es esta memoria epigenética si yo puedo de verdad
solucionarse a que la célula ya se olvide de lo que fue y que realmente se cometan hacerlo por impotente habrá un
pasado gigantesco o sea que entonces estamos todavía hemos conseguido saber que estas marcas son
importantes y que borrando podemos volver a un estado primigenio pero no es tan sencillo es mucho más complejo
vamos las conclusiones hay que seguir investigando claro bueno eso por eso siempre eso está claro y si me permiten
agua aquí el inciso de que es una cosa que como científicos como sociedad española de bioquímica y científicos
pero también como ciudadano tenemos que apoyar y este tipo de actividades también son muy importantes para que la
gente comprenda la importancia por todo lo que estamos debatiendo espero que les estemos convenciendo de que es muy importante estudiar la epigenética en
este caso que es el tema que nos ocupa hoy debemos presionar a nuestros mandatarios a nuestros políticos para
que la inversión en el sistema de investigación tanto en nuestro país como en cualquier otro país
pues sea apoyada de la forma adecuada para poder progresar tenía que otra
pequeña nota que me he hecho respecto a lo que comentábamos de esta estructura del adn porque un término que cuando yo
estudiaba y tampoco hace tanto pero bueno ya unos cuantos años solíamos hablar de él o nos hablaban del
adn basura este es un tema interesante todas estas secuencias que les contaba
al principio los genes cuando se empezó a estudiar el genoma humano nos dimos cuenta de que el porcentaje de toda la
cadena de adn que tenía información genética transmisible a una proteína es
decir algo que sabíamos que funcionaba era muy pequeña y que la mayoría del adn
pues no se sabía para qué estaba ahí y dijo no sé nada de basura no sirve para nada este término que es bastante
desafortunado y hoy sabemos que no es así porque precisamente esta arquitectura compleja que estamos
haciendo y muchas de las modificaciones y muchas de la información no para construir el ladrillo sino la
información que dice construye este ladrillo o cuando lo tiene que construir o cuánto lo tiene que construir o
durante cuánto tiempo está en en esta basura que hoy sabemos que no es
así y esto tiene también muchas implicaciones por una parte para las enfermedades porque estos cambios que
estamos hablando pueden ocurrir en zonas del adn que cuando las leemos pues parece que no son nada y tienen muchas
implicaciones evolutivas porque claro así es como la evolución me va trabajando es decir modificando no a lo
mejor la propia información sino la cuando se tienen que expresar los genes
es lo que lo que hace diferentes unas especies de otras esto es esto si queréis comentar algo al
respecto del tri dimensional del genoma pues es una nueva capa regulatoria que
se viene estudiando desde hace relativamente poco que también se considera como una capa
epigenética porque no depende de la secuencia del dna que es cómo se empaqueta el dna dentro de un núcleo es
decir cómo se dobla como no es lo mismo que bueno sabemos que es una doble hélice que es una doble hélice que se
enrolla sobre sí misma pero luego las estructuras de orden superior es
decir aquellas que tienen un grado de complejidad mayor
[Música] bueno tenemos que en cuenta que son dos metros de molécula verdad que se empaquetan en 10 nanómetros que es lo
que mide el núcleo de diámetro perdón un 100 nanómetros entonces
como se dobla esa molécula dentro del núcleo es se está viendo que es algo muy
una pregunta muy relevante y que es realmente
una cuestión que pues determina muchos
aspectos de el desarrollo del embrión o por ejemplo a un tema como la
polidactilia no a la polidactilia se ha visto que a depende hay una zona del
genoma que determina pues de cuántos dedos tenemos en la mano si esa zona del genoma se empaqueta diferente es decir
se dobla sobre sí misma de una forma aberrante se generan fenómenos como la
polidactilia y pues ya se está viendo que hay muchas pues muchas enfermedades
muchos aspectos de la biología que dependen específicamente de cómo se enrolla el dna dentro del núcleo y bueno
pues si es una área de estudio que mi laboratorio pues está explorando no si es un poco
sí porque una de las áreas más activas es estas comparaciones que se hacen entre distintas especies y estas estas
zonas volviendo un poco a las técnicas que utilizamos de una manera muy muy sencilla allí e intentando ya llegar un poco más
a un detalle como digo sin abandonarla a la parte divulgativa conocemos por ejemplo marcas específicas que hacen
cosas concretas es decir sabemos que una proteína de las que se pegan al adn de las que normalmente están pegadas al adn
dándoles parte de esta estructura primaria que yo tiene para que se organice pues hay por ejemplo dos o tres
modificaciones en esa proteína y esas modificaciones más o menos hasta donde sabemos siempre hacen lo mismo ejemplo
activar un gen cuando digo activar es hacer que algo ahora que te produce es y
hay otras que al contrario cuando están esas marcas que a lo mejor es la misma proteína pero es otra marca diferente
como si tuviera dos tapones uno en un sitio y en otro de distintos colores hace pues que no se nos expresen y
estudiando este tipo de marcas comparando unas especies con otras somos capaces de identificar regiones por
ejemplo que son importantes que están conservadas y es una de las cosas con las que se
estudian cuco cómo se como se hace en estos estudios es decir de una manera
sencilla qué tipo de técnicas utilizadas por ejemplo alejandra para saber de detectar por claro nosotros aquí vemos
el tapón de color verde pero como en un laboratorio tú puedes decir pues veo esta marca si antes de hacer un
comentario con respecto con lo que dijiste que es súper relevante nosotros
pensamos que somos especiales pero la verdad es que los mecanismos moleculares que ocurren en nosotros o en una mosca o
en una hormiga son exactamente los mismos el adn
en la bacteria en el mismo los componentes son los mismos lo único que
cambia es la organización entonces hay mucha homología a nivel
molecular en los procesos que ocurren tanto en humanos como en especies como
ratón o qué sé yo mosca y debido a eso podemos usar esos modelos
para investigar hay gente que usa gusanos hay gente que usa levadura hay
gente que usa células humanas y finalmente llegamos a hay variaciones
pero finalmente los mecanismos que ocurren en las distintas especies son los mismos ahora como estudiamos
infinidad de técnicas en mi laboratorio somos bastantes bioquímicos lo que nos gusta es purificar esto o sea a partir
de una gran cantidad de células por ejemplo y llegar a esta proteína y
caracterizar esta proteína investigar cómo funciona en el tubo ensayo si yo le
pongo más temperaturas y le pongo en más azúcar etcétera qué pasa
y una vez que uno entiende esos mecanismos muy básicos puede ir a empezar a variar los a modificar lo
bueno qué pasa si yo le cambio en la la secuencia en desde que tenga este
tubo así que cortar lo que pasa con esa proteína porque eso también nos va a ayudar a entender cómo funciona y quizás
llegar a entender una enfermedad no porque cambia utilizamos entonces métodos de
purificación en este momento debido a toda la tecnología que existe ya hay por
ejemplo secuenciación masiva lo que decías tú recién de la secuenciación del
genoma humano fue además de todo el conocimiento que nos trajo fue una un
abrir y una ayuda técnica pero impresionante y en este momento ya
podemos secuenciar el genoma completo en es barato lo podemos hacer en cualquier
momento podemos secuenciar infinidad de especies en el mismo
momento entonces las técnicas han avanzado muchísimo y ya no estudiamos solamente una
en un momento cuando yo partí mi estudio investigamos esta región pero en este
momento ya se investiga el genoma completo de las células y al mismo tiempo se estudia estas marcas a nivel
de todas las células ya no solamente en una región chiquitita sino que a nivel
general y eso también a seguir es favorecido por técnicas que parten
como investigación básica y después a una investigación a un investigador se le ocurre que se puede aplicar en otra
cosa y entonces así se van generando conocimientos nuevo y también técnicas nuevas que ayudan muchísimo no puedo
entrar en detalle porque no tienen ningún sentido general
minutillos su minuciosa y extraordinaria los que somos un poco más perezosos hacemos otras cosas
utilizamos técnicas que te permiten ver estas marcas sin necesidad de tener que purificarlo porque de verdad es un
trabajo extraordinario de tener que purifica las cosas entonces puedes utilizar una proteína que reconoce la
tapita verde y esa le pega es una cosa que es fluorescente y entonces puedes verlo en la célula en vivo en el
microscopio es la marquita cómo se mueve dentro del núcleo eso hacemos los que hacemos un poco más precios no hay que
aleja nada son técnicas complementarias son y también y en cuanto a la comparación con las especies por ejemplo
nosotros vamos a utilizado en el laboratorio para un problema de regeneración por ejemplo los peces
regeneran la retina y los mamíferos no entonces nosotros queríamos saber qué es
lo que tiene el pez que no tenemos nosotros o qué es lo que tenemos nosotros que molesta y que no debería estar en cuanto a epigenética porque
teníamos pistas de que esto podría ser un problema genético y lo que hicimos fue comparar directamente lo que es lo
que hacen las cosas epigenéticas que ocurren algunas que ocurren en el pp después del daño para que mientras va
regenerando la retina y lo que hace el ratón por ejemplo el mamífero y
buscábamos obviamente diferencias y no similitudes y encontramos algunas cosas que ocurren igual y otras que son
totalmente diferentes cientos nos vamos a poner esas claro en esas actividades epigenéticas que están haciendo si yo la
puedo modular en el laboratorio a lo mejor puede hacer que el ratón regenere la retina
y bueno obviamente hay muchas utilizas el conocimiento decía alejandro muy bien
que la ciencia la hacemos todos en conjunto con muchos laboratorios trabajando utilizas la información que
publica otra gente otros laboratorios para crecer tu propia investigación y
hacer una escalera que va conduciendo no sé si en este sentido otra cosa importante que también hemos oído y
muchas veces le ponemos apellidos a la ciencia ciencia básica ciencia aplicada y a veces por supuesto que queremos
solucionar los problemas y queremos hacerlo lo antes posible pero quizás sea equivocado centrarnos en enfocar la
ciencia en solucionar un problema la ciencia es conocimiento y el conocimiento siempre es importante y siempre va a dar un fruto mañana o
dentro de 20 años en este sentido por ejemplo vuestras investigaciones que pueden parecer que son muy básicas o que
no llevan a ningún sitio o la pregunta que a todos los científicos nos suelen hacer y eso que tú haces para qué sirve sirve para aprender y aprender siempre
está bien y por supuesto aprender cosas de detalle de los que estamos hablando pues pues mucho más querías fabiana
mencionar porque el tema de los anticuerpos a mí me parece importante porque a lo mejor mucha gente ha oído
hablar de anticuerpos como algo de nuestra defensa pero es una de las herramientas más fundamentales que usamos todos los científicos en el
laboratorio a estudiar las marcas epigenéticas por ejemplo si hay una marca amarilla
indicar que un gen se va a activar hay anticuerpos específicos para la marca amarilla la marca roja la marca verde
entonces se se incuban se ponen en distintos ambientes y
después se incuban con esos anticuerpos se precipita se purifica y se identifica que gen está o qué sector del adn está
marcado con esa marca que vamos a saber que se va a activar o se va a inhibir entonces este identificando esas marcas
con anticuerpos específicos de las marcas identificamos qué marcas hay identificamos que hay distintas marcas
de acuerdo en qué en qué ambiente esté esa célula si el tema de los anticuerpos como herramientas de investigaciones es
fascinante y es un ejemplo más de cómo algo que la naturaleza digamos ha producido para un fin que es defendernos
de las infecciones porque recuerdan que un anticuerpo es una molécula es una
molécula que reconoce a otra básicamente entonces normalmente lo que lo que hacen es reconocer por ejemplo
bacterias virus que nos infectan para eliminarlos esa es su función natural si nosotros ahora desarrollamos anticuerpos
contra el tapón amarillo pues somos capaces de las células en el tubo de ensayo usar
estos anticuerpos como una herramienta que nos permite saber y identificar dónde están estos anticuerpos otro tema
que está muy dado muy de moda ahora a lo mejor han oído es el tema de la edición genómica el cris per es una técnica que
lo mismo es una tecnología un ejemplo que unas bacterias desarrollaron para defenderse y que nosotros ahora hemos
utilizado para precisamente cambiar la secuencia del adn de una manera muy
mucho más sencilla de lo que de lo que ocurría hasta hasta ahora
a mí esto me parece tremendamente relevante porque de nuevo con la idea de
que nosotros tenemos que hacer investigación que sea aplicada o que tenga la investigación no necesariamente
nos va a llevar a eso o sea puede que estemos pensando de una manera que no nos va a llevar a ninguna a ninguna parte sin embargo investigaciones como
lo que están mencionando con jose de crisis percas que en la edición higiénica que se está haciendo ahora fueron investigadoras mujeres que
estaban interesadas en invertir en entender cómo la bacteria se defiende o
sea si yo hubiese dicho eso no sé 20 años atrás quizá había medir tubería ti
qué te importa o sea para que no pero sin embargo ese conocimiento está
y bueno después alguien se le ocurrió a espero esto lo podemos utilizar y yo
creo que va a ser una de las revoluciones medicina porque es probable que podamos después editar nuestro genoma no ahora en un tiempo más pero es
una cosa que lo más probable va a pasar y va a revolucionar la medicina entonces uno no sabe por donde la aplicación del
conocimiento va a ocurrir y entonces enfocar la investigación en solamente
aplicación muchos gobiernos lamentablemente en mi país ocurre muchos políticos quieren ver efectos o
resultados inmediatos y la ciencia no es así no es así muy lejos puede que hay un
descubrimiento que se hizo veinte años atrás y que la persona que lo hizo nunca se le ocurrió pero a la persona que lo
leyó a mira esto cómo publicar estas cosas y ahí se genera la aplicación pero
uno no hace una investigación basada en aplicación yo creo que eso es un error que mucha gente tiene y por ahí no va o
sea no podemos no existe la ciencia aplicada no existe es la generación de
conocimiento y en algún momento eso va a llevar aplicación es algo que uno tiene que entender y que
darle los tiempos a los científicos para que investiguen no son los mismos tiempos los políticos que quieren
reelección y entonces quieren resultados de ciencia tiene otros tiempos que son
hermosos no porque nos da tiempo para investigar pero no es la
investigación es generación de conocimiento con la potencialidad de
utilizarla
…
quiero contarte la historia de Anne y Marie, dos gemelas idénticas nacidas
en el mismo lugar, criadas en la misma casa y, como gemelas que eran, increíblemente
parecidas.
Sin embargo, esas similitudes no duraron para siempre:
A los 30 años, Marie fue diagnosticada con trastorno bipolar, mientras que Anne siguió
manteniendo un buen estado de salud mental.
No obstante, a los 40 años Anne fue diagnosticada de enfermedad cardíaca grave, mientras que
Marie se mantuvo en buena forma física hasta muy avanzada edad.
No solo eso sino que, a medida que pasaban los años, las hermanas gemelas se iban pareciendo
cada vez menos.
Este ejemplo concreto es ficticio, pero como este, existen muchísimos casos reales que
muestran cómo gemelos monocigóticos, es decir, gemelos que surgieron del mismo óvulo
fecundado y que por tanto comparten exactamente la misma información genética, han sufrido
enfermedades muy diferentes y han ido sumando diferencias a medida que pasaban los años.
PERO si nuestra información genética realmente dicta lo que somos y cómo funcionamos, ¿Cómo
pueden ser tan diferentes si comparten el mismo ADN?
Hoy en la Hiperactina, hablamos de EPIGENÉTICA.
Durante mucho tiempo y en varios vídeos del canal hemos dicho que el ADN de nuestras células
contiene la información genética, que define quiénes somos, cuáles son nuestras características.
Y esto es en cierto modo verdad: es el motivo por el que una alteración genética, por
ejemplo un cambio en la secuencia de información que conforma el ADN puede dar lugar a enfermedades
tan conocidas como el cáncer, la fibrosis quística o el daltonismo.
Sin embargo, aquello que dicta lo que eres, cómo funciona tu cuerpo e incluso las enfermedades
que puedes desarrollar en un momento concreto no solo viene dado por tu ADN, sino por la
forma en la que este se lee.
A ver, dentro de lo que es la larga secuencia del ADN, los «genes» son fragmentos dentro
de esa secuencia que contienen la información para sintetizar las proteínas, esas moléculas
que necesita la célula para funcionar.
Concretamente, cada gen tiene la información para sintetizar un tipo de proteína. pues
cuando un gen es “leído” y da lugar a su proteína, decimos que ese gen se está
«expresando».
Es decir, el gen está activo, está teniendo un efecto en la célula.
Y la gracia de esto es que esta expresión de genes va variando en función del momento,
es decir, la célula activa y desactiva los genes según lo que necesite en cada momento,
lo cual es súper importante porque le permite adaptarse a los cambios constantes en su entorno.
Por ejemplo, imagínate que estás cocinando y te quemas con la sartén.
En otras palabras (más biomédicas), se ha producido un daño en un tejido en el que
han muerto células que deberán ser reemplazadas.
Para reparar ese tejido dañado, las células de alrededor de la herida deberán dividirse
para dar lugar a nuevas células, es decir, tendrán que activar los genes involucrados
en la división celular.
O sea, que la célula necesita encender y apagar genes según le convenga, pero ¿Cómo
hace esto?
¿Cómo se consigue activar y desactivar un gen a tu antojo?
Pues precisamente a través de la llamada epigenética.
Epigenética significa “por encima de la genética”, y básicamente consiste en añadir
o quitar pequeñas moléculas químicas en zonas concretas capaces de activar o desactivar
genes.
Es decir, son marcas químicas que cambian la forma en la que se lee el ADN, pero sin
alterar su secuencia genética, que en el fondo sigue siendo la misma.
Por ejemplo, imagina la frase “No está mal la homeopatía”.
Si te digo esto, seguramente entiendas que no tengo una mala opinión sobre la homeopatía,
mientras que si le añado algunos signos de puntuación… el significado de la frase
cambia totalmente.
Haciendo un símil con el ADN, podríamos decir que el ADN vendrían a ser las palabras
de un texto, y la epigenética serían los signos de puntuación que harían que se leyese
de una forma u otra.
Vale, todo esto suena muy abstracto, pero yendo a la chicha, a la biomedicina, ¿Qué
significa exactamente?
¿Qué le hace la epigenética a nuestros genes para cambiar la forma en la que se expresan?
Pues prepárate para ver una vez más por qué nuestro cuerpo es increíblemente complejo…
y fascinante.
Como hemos dicho, las modificaciones epigenéticas consisten en añadir pequeñas moléculas
químicas en sitios muy bien pensados del ADN.
En este vídeo, vamos a ver concretamente dos formas de cambiar la expresión de un
gen: alterando directamente el ADN o alterando las proteínas que empaquetan el ADN.
Empecemos por esta última.
Una de las cosas que más me fascinan del cuerpo humano es la burrísima longitud que
tiene el ADN de nuestras células: dentro de cada una de nuestras microscópicas células
se guarda un total de 2 metros de ADN.
¿Cómo salen las cuentas?
Pues eso es así porque el ADN no está todo desparramado por la célula, sino que se encuentra
ultra empaquetado de forma muy bien estructurada y diseñada gracias a unas proteínas llamadas
histonas, sobre las que se enrolla el ADN para empaquetarse.
Si entendemos el ADN como una secuencia de información genética que la célula necesita
“leer”, es fácil imaginar que si ese ADN está muy empaquetado, este será ilegible.
Básicamente porque las proteínas que se encargan de “leer” el ADN no podrán acceder a él correctamente.
Por tanto, los genes que se encuentren en zonas del ADN muy empaquetadas estarán inaccesibles y por tanto esos genes estarán “apagados”, no se van a expresar.
¿Qué debemos hacer si queremos activar esos genes?
Pues siguiendo la misma lógica: desenrollar el ADN.
Cuando las histonas desenrollan el ADN, esa información vuelve a estar accesible y sus genes pueden e: expresarse están “activos”… papi.
¿Y cómo controla una célula que las histonas desenrollen o enrollen los fragmentos de ADN que le interesan? Pues a través de la epigenética.
La célula añadirá pequeñas moléculas químicas a las histonas que cambiarán su estructura y por tanto el empaquetamiento del ADN.
Aunque existen otros ejemplos, una de las moléculas químicas que pueden hacer esto es el llamado grupo acetilo: cuando se añade un grupo acetilo a una histona, esta cambia su estructura de forma que el empaquetamiento se relaja, y el ADN pasa a estar accesible para leerse.
Pero la epigenética es reversible: también podemos hacer lo contrario, quitar ese grupo
acetilo, de forma que la histona, vuelve a cambiar su estructura, compacta de nuevo el
ADN y este ya no se puede leer.
Y este juego tan tonto de empaquetar y desempaquetar el ADN es un ejemplo de cómo las células
pueden tener algunos genes activos mientras que otros están silenciados.
Algo que permitió, entre otras cosas, que nuestras células se especializasen en funciones
muy concretas, dando lugar a distintos tipos celulares.
Aquí viene un dato mindblowing de la biomedicina: todas las células de tu cuerpo tienen el
mismo ADN, los mismos genes, ya sea una célula hepática, una neurona o una célula de tu
retina.
WTF, ¿Cómo es posible si son tan diferentes?
La cosa es que gracias a la epigenética que permite modificar la estructura de las histonas
y cómo se empaqueta y se lee ese ADN, cada célula tendrá activos únicamente los genes
que le interesen: por ejemplo, una célula del hígado expresará genes que le permitan
metabolizar ciertos fármacos, pero no le interesarán los genes que nos permitan ver
con nuestros ojos, mientras que en una célula de la retina pasará lo contrario.
En resumen: la modificación de las histonas es una forma útil de cambiar la expresión
de los genes a nuestro antojo, pero existe una forma de epigenética más directa, que
en lugar de afectar a las proteínas que empaquetan el ADN, afecta directamente al ADN.
La forma más sencilla de epigenética y también la más estudiada, es la llamada metilación
del ADN.
Se llama así porque consiste en añadir una molécula química llamada “grupo metilo”
a las bases nitrogenadas del ADN, concretamente a las citosinas, para que veáis hasta qué
punto es todo ultra específico.
Este grupo metilo, ahora unido a la molécula de ADN, va a impedir que se le adhieran las
proteínas necesarias para leer el gen en concreto, con lo que este quedará inactivado.
Sería algo así como si tuviésemos un libro (que representaría todo el ADN, toda la información
genética) y esta página fuera un gen.
Si le pusiéramos una pegatina enorme, estaríamos impidiendo que la persona encargada de leer
este libro pudiese hacerlo, bloqueando la lectura de esa página.
Por suerte, como hemos comentado, la epigenética es un proceso reversible: igual que podemos
añadir el grupo metilo que “bloquea” el gen, también existe el proceso contrario,
la desmetilación, en la cual se retira ese grupo metilo y el gen se activa.
Lo mismo pasaba con las histonas que hemos visto: añadir un grupo acetilo desenrollaba
el ADN y activaba el gen, mientras que quitarlo lo volvía a enrollar, inactivando el gen.
O sea, que estamos diciendo que la epigenética es capaz de, partiendo de una misma información
genética de un mismo ADN, cambiar la forma en la que ESTE SE LEE simplemente añadiendo
o quitando ciertas moléculas.
Y ESTO nos lleva de vuelta a la historia de antes, al ejemplo de las gemelas: por mucho
que dos personas nazcan con la misma información genética, nunca van a ser iguales, porque
su epigenética va a ser distinta.
Nunca estarán expresando los mismos genes en cada momento de su vida.
Esto es lo que han visto en multitud de estudios al comparar la metilación del ADN o de la
acetilación de las histonas en las células de gemelos idénticos y ver que efectivamente
existían diferencias epigenéticas entre ellos ya desde su infancia.
No solo eso sino que cuanto más mayores son los gemelos, menos se parecen.
Y no hablamos solo de diferencias físicas, evidentemente, sino también en el desarrollo
de ciertas enfermedades: se ha visto que los gemelos idénticos presentan de forma distinta
enfermedades como la diabetes tipo 1, la diabetes tipo 2, enfermedades autoinmunes, enfermedades
mentales como el trastorno bipolar y la depresión, o distintos tipos de cáncer.
Por ejemplo, en este estudio se compararon gemelas en las que una de ellas había sido
diagnosticada de cáncer de mama, y se observó que aquella mujer que desarrollaba el cáncer
presentaba una metilación exagerada en un gen concreto: el DOK7.
Esto nos puede servir por ejemplo para diagnosticar un cáncer a tiempo, es decir, DOK7 se podría
utilizar como “biomarcador” que indicase la presencia de un cáncer de mama en el cuerpo.
Y después de todo esto, la pregunta estrella es: ¿Por qué esas diferencias?
¿Qué condiciona que la epigenética evolucione de forma distinta en dos personas a priori
idénticas?
Pues aquí viene lo interesante.
Precisamente la epigenética podría explicar la influencia que tiene el entorno en nuestras
características o nuestra salud, es decir, el por qué cosas como la dieta, hacer o no
ejercicio, estar expuestos a ciertas sustancias o tener ciertas infecciones, pueden cambiar
la forma en la que se comportan nuestros genes y por tanto influir en la probabilidad de
padecer ciertas enfermedades.
Pero es que la cosa va más allá: como hemos dicho la epigenética es reversible,
con lo que si conocemos qué cambios epigenéticos se encuentran en determinadas enfermedades
como el cáncer, podríamos llegar a desarrollar fármacos que actúen específicamente sobre
esos cambios para revertir la enfermedad, cosa que ya se está haciendo hoy en día.
O sea, que hemos pasado de soñar con algo tan GRANDE como la EDICIÓN GENÉTICA especialmente
gracias a técnicas tan recientes como el CRISPR, a soñar con algo incluso más específico:
la edición EPIGENÉTICA.
La epigenética es un tema fascinante porque nos enseña una vez más lo maravillosamente
complejo que es nuestro organismo, y cómo por mucho que logremos hasta secuenciar toda
la información genética que se esconde en nuestro ADN, hay algo incluso más allá que
hasta hace unos pocos años ni siquiera hubiéramos imaginado.
Me recuerda a la frase de Manel Esteller, Director del Instituto de Investigación Josep Carreras e investigador de la epigenética del cáncer en la que dice: «Se decía que el ADN era el libro de la vida, pero le faltaban las comas, los puntos, es decir, la epigenética».
Me he dejado algunos puntos como la herencia de la epigenética,
EPIGETICA
Cada dia , pero cada dia un nuevo conocimiento científico, modifica a lo anterior.
Y no es posible ignoraralo porque en ellos esta la vida.
Shinya Yamanaka 2006, John B. Gurdon reprogramación celular
LA APARICION DE LA EPIGENETICA, ES UNA REVOLUCION COMO LO SON CADA DIA, LAS NUEVAS APARICIONES.
Las dificultades del conocimiento vienen porporcionada, porque los nuevos hallazgos, desmontan totalmente lo anterior.
Esto es una copia de una conferencia Hispano Americana sobre epigenetica
FÉLIX GOÑI catedrático de bioquímica y biología molecular de la universidad del país vasco y el actual presidente de la SEP y también del doctor JUAN JOSÉ SANZ tenemos hoy aquí como moderador científico titular en el centro nacional de biotecnología del consejo superior de investigaciones de américa
Nuestros participantes son la doctora LORENA AGUILAR del instituto de investigaciones biomédicas de Mexico la doctora MÓNICA LAMAS del centro de investigación y estudios avanzados también de MEXICO la doctora ALEJANDRA LOYOLA de la fundación ciencia y vida de CHILE y la doctora MARÍA FABIANA DRINK coach de la universidad nacional de Rosario Argentina
Todos han oído hablar del ADN y de nuestros genes.
Esto es un modelo de la doble hélice de ADN transporta la información genética que está contenida en esta molécula y está dentro de todas nuestras células y determina como somos como son nuestras características morfológicas y fisiológicas y muchas veces también qué tipo de
enfermedades vamos a padecer.
Si la desarrollamos el ADN vemos que es una secuencia de componentes químicos cuatro diferentes que van formando una lista y cada uno de nuestros genes, nuestras información genética está compuesta por una combinación diferente de todas estas letras
Hasta hace poco pensábamos que ahí estaba todo Pero hay algo más y a esta molécula de ácido nucleico que forman nuestros genes del ADN se pegan proteínas que son las que hacen nuestra. funciones.
Estas moléculas, se pegan y hacen que el ADN transmita esa información
a otras moléculas.
De forma que estas proteínas se pegan a la cadena de ADN aquí manejan como funcióna la cadena de ADN y cada una de sus secuencias.
Son como etiquetas de modificaciones químicas.
LA EPIGENÉTICA LO QUE ESTÁ POR ENCIMA DE LA GENÉTICA
La genética es la secuencia de nuestros genes, la epigenética es lo que se pone sobre ellos regulandolos.
El tipo de etiquetas que a veces tiene también el propio ADN va a hacer que se
comporte de una manera o de otra y que algunos genes se activen funcionen y otros genes no funcionen correctamente.
Estas etiquetas se colocan dentro de las moléculas y nos hacen entender cómo funciona la salud.
Muchas otras cosas son vitales para el funcionamiento de la células no solo en humanos, sino también en animales y plantas.
Y asi podemos entender e incluso manipular las características de un ser vivo.
John Gurdon y Kim premios Nobel observaron hace 75 años,
que las distintas células que componen los organismos, tales como neuronas, linfocitos, células hepáticas células tienen la misma información genética el mismo ADN , sin embargo son tremendamente diferentes .
En los 50 se pensaba que la diferenciación celular se debía a que la célula perdía información genética, y entonces una célula muscular iba a tener
información genética distinta a una la neurona porque perdía
información y eso le daba la diferencia, pero en ese año se hizo el
primer planeamiento el primer ciclo no una rana desde se sacó desde una célula que ya pertenecía a una rana adulta y se pudo hacer una nueva rana lo que significa la importancia de ese experimento fue que se demostró que esa célula que estaba diferenciada en una rana adulta contenía toda la información de una que podía generar una nueva rana por lo tanto no había pérdida de información entonces. 
La siguiente idea que una célula neuronal una célula muscular tiene la
misma información genética pero expresa distintos genes y eso se regula a nivel genético entonces, ese es un experimento que fue tremendamente importante para definir que hay algo más en el ADN que permite entonces tener distinta información en distintas células.
Estas modificaciones pueden estar tanto en la propia secuencia del ADN como en las proteínas
que sintetiza
Estas modificaciones estas marcas que se ponen en el ADN o en las
proteínas asociadas lo que hacen es que cambian la arquitectura del DNA vale
entonces digamos la imagen muy sencilla de entender
Es que el ADN está formado
por ladrillos, cuatro ladrillos que son idénticos y que se repiten en un orden determinado
y una serie de grupos químicos que se repiten en un orden determinado uno con unos ladrillos puede hacer a un
apartamentito a las afueras de Madrid o el palacio de Linares y esto es lo
que hacen estas marcas se fijen en la epigenética se refiere justamente a esto a la arquitectura del
DNA cómo está organizado así lo que tienes en una zona determinada es el apartamento a las afueras de Madrid o en
el palacio de Linares y tienes una neurona funcionando a todo lo que da entonces.
Durante mucho tiempo nosotros estuvimos
los biólogos moleculares con mucho detalle
justamente como de mención a Alejandra cómo se expresan los genes como hacer que un gen se produzca cómo se
produce la proteína y era como si viéramos durante mucho tiempo una parte muy pequeña de algo y
de repente alguien hubiera quitado una cortina y hubiera visto que hay toda una historia detrás inmensa que no
considerábamos y que entonces ahora tenemos que considerar porque la imagen es diferente no es lo mismo estar
estudiando una parte de un dedo de una estatua y luego quitar la cortina y ver que lo que tienes es el David de Miguel A
ngel.
Esto es lo que ha hecho la epigenética y estos cambios son los que tenemos que estudiar. sobre lo que estudiábamos antes .
Una nueva visión esta parte del
iceberg que está por debajo del agua esto es lo que estamos estudiando y esto es lo que está afectando absolutamente a cómo vemos las enfermedades cómo entendemos el funcionamiento de las células etc porque estos cambios que estamos mencionando que son muy muy
variados por una parte es decir hay muchas de estas etiquetas muchas marcas pero además son muy dinámicos.
Una de las características más importantes
que realmente realzan la relevancia de lo que es la epigenética es precisamente
eso lo que es un mecanismo que tiene gran plasticidad y gran dinamismo y es
reversible porque lo que es el genoma que nos enseñaban y lo que nos enseñaban antes.
Con esta molécula el genoma
posibilita que el DNA funcione y se generen los procesos que el propio DNA
debe llevar a cabo son estos mecanismos epigenéticos porque ellos son los que tienen movilidad plasticidad
cambian a dependiendo del tipo celular dependiendo de los estímulos los
estímulos ambientales son los que permiten que el DNA ejerza
su función adecuada en cada momento en cada tipo de celular y en respuesta a cada situación ambiental en el en la
cual se encuentra la célula y quizás la
característica que yo encuentro más atrayente para para dedicarme precisamente al estudio de la tijera gen
ética sí creo que esto que menciona Lorena.
Es muy importante el tema de las
mutaciones y el tema de la plasticidad el epigenética por supuesto que la secuencia del ADN puede cambiar,
puede haber un error, nosotros estamos preparados para que este modelo sea la doble hélice está pensada precisamente
para que se copie a sí misma cada una de las dos cadenas se separan y cada una
sirve de molde para que la otra cadena como si fuera un espejo se copie sobre ella.
Esto es un proceso
físico y puede haber errores y las células están
preparadas para corregir esos errores pero a veces no pueden con ello entonces hay factores que son capaces de
introducir mutaciones en este ADN no ocurren porque
estamos preparados para ello y si ocurren pueden dar lugar a enfermedades.
Hay factores como puede ser por ejemplo la luz ultravioleta del sol
que puede dañar el ADN y por eso tenemos que protegernos porque esas mutaciones se quedan fijadas en el ADN una vez que
ya han sucedido y se transmiten de una célula a la siguiente en cambio todo este dinamismo de la epigenética todo
de las etiquetas moleculares también está muy influido por cosas por factores
ambientales que hasta ahora no éramos muy conscientes y que ahora estamos aprendiendo que no solamente por ejemplo
en este caso pues la luz en sí misma sino que la alimentación la polución la
contaminación hay muchos factores externos que pueden tener un impacto
Hay marcas que dicen que un determinado gen se va a expresar o que un determinado gen no se va a expresar y entonces
nosotros lo que hacemos es estudiar esas marcas para saber este si un gen determinado se va a expresar o no.
En las
plantas por ejemplo se ha visto qué no florecen en el
invierno y se ha visto que está implicada la epigenética en eso se ayuna hay una proteína que se expresa en
determinado momento durante el invierno y hace que no se desencadene la floración y cuando cambia el ambiente
cambia la temperatura cambian determinadas condiciones o las condiciones de luz éste se produce una
marca epigenética que hace que una proteína se exprese y entonces la planta florezca y lo maravilloso que se ha
visto también de estas marcas es que por ejemplo en las plantas se heredan o sea es una memoria que si una determinada
planta por ejemplo se vivió en un determinado ambiente y tiene determinadas condiciones
de generaciones de las plantas esas marcas se van manteniendo o sea no sólo se van manteniendo la secuencia del ADN
sino que por un tiempo hasta que se marque se desmarquen esas marcas también se heredan entonces es esto de un área
que nos ha cambiado un poco este el paradigma de cómo estudiar algunos aspectos cómo abordar algunos
estudios en bioquímica y en biología molecular que nos ha impactado a todos los científicos pero sí que me gustaría
añadir qué esto qué ocurre en las plantas ocurre también en los humanos y qué es una de
las cosas que justamente la epigenética incide hablando de la pregunta que nos
hacemos hoy de cómo incide sobre nuestra salud lo que hacemos nosotros hoy puede afectar a nuestros nietos y por ejemplo
y esto ha habido experimentos hechos en gente que pasó hambre durante la segunda guerra mundial la famosa hambruna en
Holanda hay gente que pasa mucha hambre por el racionamiento de la alimentación los nietos de esta gente hoy en día
podemos seguir las marcas epigenéticas que causó aquella condición y los nietos todavía tienen marcas
epigenéticas en su ADN, o sea que su ADN funciona de una manera particular eso es histórico así fueron los primeros
experimentos que se hicieron sobre estas era de habilidad en el humano y esto implica que hoy en día lo que nosotros
hacemos lo que bebemos lo que comemos el atasco de la m-30 en vivir en la ciudad de México en fin estas cosas
influyen sobre mí epigenoma l y el de nuestros descendientes también y
es una cosa más de las que tenemos que tener en cuenta que antes no
conocíamos esto que mencionas Mónica es muy interesante porque ciertamente hasta
eso significa que modificaciones de nuestro ambiente son capaces de pasar a
generaciones posteriores y eso digamos que era una va en contra del dogma de la
de la herencia genética pero hoy en día efectivamente hay experimentos que demuestran que estas marcas epigenéticas
algunas la mayoría son muy dinámicas son lo que llamamos reversibles son de quita
y pon pero hay otras que son tan estables que incluso pueden pasar las generaciones es cosas y verdad hay experimentos el que has mencionado y
otros en el laboratorio en el que esto se ha demostrado en el en el yo conozco algunos en particular en el área de la
psiquiatría en la que justamente hay una idea de que digamos estas marcas
epigenéticas al principio me digan por ejemplo en el mundo de las adicciones
Las adicciones causan cambios en el epigenoma de la gente, son de estas marcas epigenéticas de la persona adicta
y estas marcas pueden ser reversibles durante un tiempo pero si se mantienen y se mantienen al final se hacen estables
y esas marcas epigenéticas se pueden pasar a la progenie incluso la progenie
la puede pasar a la siguiente progenie efectivamente esto ya está comprobado está comprobado que tenemos un nivel
adicional ya no es sólo para nosotros sino para nuestros hijos nietos etc
Querria que me comentara si alguna en
algún caso concreto de enfermedades por ejemplo en humanos que tengan una base
genética y que esté muy claro muy estudiado alguien tiene a lo mejor incluso trabajáis directamente en vuestros laboratorios en este campo
bueno yo creo que es un campo que el campo de la ficción ética es un campo relativamente nuevo que
no permite o sea es muy complejo porque hay muchos factores que
influencian hay mucha maquinaria molecular implicada muchas proteínas distintas muchas integraciones redes
genéticas que los coordinan y por tanto el entenderlo pues todavía estamos en
ello en lo que son lo que se denominan enfermedades complejas como por ejemplo:
El tema de la obesidad o el desarrollo de diabetes a enfermedades como
enfermedades neuropsiquiátricas que tienen muchos factores que convergen
para que se dé la enfermedad pues tiene un componente genético muy fuerte pero
es de momento nos está resultando muy difícil discernir digamos encontrar el
elixir hola e la diana molecular que nos va a permitir
resolver estos problemas pero algunos avances se hacen por ejemplo en cáncer algunos tipos de cáncer lo que serían los tratamientos para curarlos pues hay algunos tipos de tratamiento epigenéticos
que permiten alguna de la máquina molecular implicada en la en el control del epigenoma y pues han probado efectivos para el tratamiento de ciertos cánceres de ciertos cánceres muy concretos y bueno yo creo que ahí el campo es donde quizás esté más avanzado La mayoría si no todas las enfermedades finalmente se reducen a que hay un cambio en la expresión de algunos factores y eso está regulado por la parte genética y por la parte de genética yo diría que los años noventa la investigación estuvo centrada en identificar o sea purificar desde una célula que crece en cultivo llegar hasta la proteína que producía la función
El hilo es buscar qué proteína ponía esta tapita verde acá y entonces fueron muchísimos años décadas donde se hizo eso muchísimos laboratorios la ciencia se hace en conjunto y nosotros tenemos mucho mucha comunicación entre los científicos publicamos nuestros datos entonces ustedes pueden buscarlo es disponible una vez que se identificaron estas proteínas que son tremendamente relevantes
Los científicos nos empezamos a dar cuenta que muchas de esas proteínas estaban montadas en algunas enfermedades la mayoría de las proteínas que van a producir la tapita verde la tapita amarilla roja están mutadas en muchas enfermedades son diferentes o funcionan más o funcionan menos o no funcionan
Enfermedades como el cáncer son enfermedades multifactoriales no se debe a una modificación a una a una falla sino que son muchas entonces es difícil definir que una proteína sea responsable hay enfermedades donde si uno puede decir un factor. La hemofilia por ejemplo se debe a que el factor 8 no se produce y eso es porque hay un problema a nivel de ADN puede ser o también puede ser un problema a nivel de la regulación de como ese factor se expresa y la pregunta se me falta algún ejemplo concreto y lo que está quedando claro es que los mecanismos son tan complejos son multifactoriales son multifactoriales hay casos de enfermedades monogénicas que es como nos referimos a ellas que un único gen o una única un único problema en un factor concreto causan la enfermedad y puede ser una enfermedad gravísima si lo que está modificado lo que no funciona bien es muy importante pero normalmente las enfermedades sobre todo las enfermedades más prevalentes las que afectan a la mayoría de la población suelen tener origen es multifactorial es entonces muchas veces es muy difícil acotar cuál es el problema y estamos hablando a lo mejor simplemente de la secuencia del ADN si ya metemos todo el tema epigenético es complicado pero es que es justamente ahí, es justamente porque hubo un momento que todos recordarán a veces la ciencia salta del laboratorio a la calle y uno empieza a ver en los periódicos el intermediario que se habla de genes y que hoy en
el gimnasio te hacen el análisis genómico para ver si vas a poder subir tres escaleras o no en fin y hubo un momento en el que todos pensábamos que el DNA era definitivo o sea que tú nacieras con un DNA y esa era tu destino incluso había una película no sé si recuerdan llamaba guataca en o que el de línea fija va a tu destino en la vida y ya no podía ser astronauta si no tenías un cierto DNA y justamente había preguntas en ese
entonces en este concepto por eso se lanzó el proyecto del genoma humano. Vamos a leer el genoma humano entero vamos a ver todos los ladrillos que constituyen el genoma humano y lo vamos a entender y vamos a solucionar todo y vamos a entender todas las enfermedades que no se conocían porque vamos a encontrar todos estos genes que no están funcionando y entonces se resuelve el genoma humano se lee y diez años después seguimos más o menos exactamente en la misma posición no entendemos nada ni sabemos nada de estas enfermedades esto es justamente lo más interesante en los laboratorios cuando un experimento no sale como uno espera, entonces te tiene que poner a pensar y entonces en este caso todas esas preguntas que no se podían resolver porque dos gemelos que tienen exactamente el mismo DNA el mismo DNA no
son iguales y uno a los veinte años desarrolla esquizofrenia y el otro no porque no hemos conocido más solución a enfermedades no después de haber leído todo el genoma humano y habernos gastado millones de dólares y porque hemos avanzado muy poco en el conocimiento pues porque las cosas no iban por ahí no rascó y cómo y cómo era esta historia mágica de no el ambiente interfiere en el DNA.
Porque un gemelo es esquizofrénico, es el ambiente pero no teníamos ni la más remota idea de cómo el ambiente intervenía
Todo eso es la epigenética como el ambiente interfiere como ya en el genoma nos dicta nuestro futuro porque el epigenoma como bien dicen todas mis compañeras se puede cambiar y si tú puedes afectar tu epigenoma y puedes cambiar el destino de esto y en las enfermedades pues en particular mucho esfuerzo realizado para encontrar un gen que fuera responsable de una enfermedad estudiando a las familias el afectado la familia a la abuela la bisabuela que viene de la argentina el otro sector mont y no se encontraban los genes porque porque no estaba en los genes estaba en la epigenética estaba en la arquitectura estaba en estas marcas que hacen que el genoma sea diferente
La epigenética ha venido a cambiarnos la perspectiva la epigenética es un campo muy nuevo no entonces pues todavía muchas cosas por describir y por descubrir y creo que te va a dar muchos más frutos de los que
está dando y todavía hay muchas cosas que podemos entender y que nos van a ayudar a comprender ciertas enfermedades de relevancia mundial ahora mismo en esa misma línea
Particularmente de los gemelos que es el paradigma de las personas genéticamente idénticas y es así y muchas veces o el argumento que se ha utilizado históricamente para defender esto es gemelos a lo mejor que son separados y luego son iguales en aficiones en comportamental pero tú has mencionado el caso contrario que efectivamente es lo que nos estamos dando cuenta casos en los que son idénticamente genéticos genéticamente igual pero por la epigenética en este caso han cambiado y en ese sentido creo que también hay estudios que hablan de una genética de la edad es decir la edad el envejecimiento o cómo vamos avanzando a lo largo de la vida parece ser que hay marcas concretas identificadas que caracterizan esto creo que eso es así verdad alguien quiere comentar algo bueno sí o sabe que es así así es claro que todavía no sabemos exactamente desgraciadamente así es que tenemos marcas verdad pero no sabemos cómo eliminarlas digamos por lo menos en mamíferos en plantas cómo está el asunto el ambiente cambió muchísimo el epigenoma las marcas que se ven en distintos ambientes en plantas que tienen el mismo adn es osea cambia muchísimo de acuerdo a si la planta crece por ejemplo con más tiempo de luz con menos tiempo de luz se ven marcas distintas y se expresan genes distintos y el ADN es el mismo siendo a esto que contabas hay un experimento también clásico con ratones este en los cuales a las madres tienen a los ratones con el mismo este mismo material genético pero la alimentación de la madre durante el embarazo influye en que el ratón salga distinto y salga con por ejemplo este exprese una determinada enfermedad o no y en la alimentación de la madre que le transmite al hijo y salen distintos la descendencia de acuerdo a ese proceso sea que el ambiente la alimentación donde nos movemos influye muchísimo en estas marcas influye muchísimo después en lo que se va a expresar.
Cuando somos embriones y las células empiezan a diferenciarse se van añadiendo distintas marcas para que una célula sea de una manera o de otra ahora hemos aprendido mecanismos para borrar
esas marcas y volver hacia atrás de forma que una célula adulta sea capaz de volver hacia un estado pluripotente esto significa que esa célula ahora la podemos convertir otra vez en otra célula diferente porque
estas marcas que se han ido añadiendo más o menos conseguimos borrarlas y ahora utilizando un cóctel diferente les convertimos en otra cosa .
El edil es del premio nobel de Yamanaka por este proceso por el que introducía cuatro genes en una célula y la convertía en una célula primigenia digamos una célula capaz de dar lugar a todas las células del organismo le dieron el premio nobel y tropecientos mil laboratorios en el mundo empezaron a tratar de justamente utilizar esta técnica para tener células una fuente ilimitada de células para poder generar con las células que a mí me hicieran falta que produjeran insulina o células de neuronas para la enfermedad de Parkinson.
Muchos laboratorios y de repente se empezaron a dar cuenta de que estas células si hacían lo que querías que hicieran durante un tiempo y luego dejaban de hacerlo y volvían a su estado original más o menos y alguien en estado muy poético dijo estas células tienen memoria epigenética.
El propio Yamanaka después de que le dieron el premio nobel dijo bueno sí claro esto es lo que llevo descubrió muy importante pero claro tiene también aquellos sus cursos sus problemas vamos el mensaje es por supuesto el avance es impresionante y es un poco lo que tú decías al principio se generan nuevas preguntas qué es esta memoria epigenética si yo puedo de verdad solucionarse a que la célula ya se olvide de lo que fue y que realmente se cometan hacerlo por impotente habrá un pasado gigantesco o sea que todavía hemos conseguido saber que estas marcas son importantes y que borrando podemos volver a un estado primigenio pero no es tan sencillo es mucho más complejo.
Al principio los genes cuando se empezó a estudiar el genoma humano nos dimos cuenta de que el porcentaje de toda la cadena de ADN que tenía información genética transmisible a una proteína es
decir algo que sabíamos que funcionaba era muy pequeña y que la mayoría del ADN pues no se sabía para qué estaba ahí y dijo no sé nada de basura no sirve para nada este término que es bastante
desafortunado .
Hoy sabemos que puede ocurrir en zonas del ADN que cuando las leemos pues parece que no son nada y tienen muchas
implicaciones evolutivas porque claro así es como la evolución va trabajando es decir modificando no a lo
mejor la propia información sino la cuándo se tienen que expresar los genes
es lo que lo que hace diferentes unas especies de otras.
Esto
es el aspecto l tridimensional del genoma pues es una nueva capa regulatoria que
se viene estudiando desde hace relativamente poco que también se considera como una capa
epigenética porque no depende de la secuencia del DNA , es cómo se empaqueta el DNA dentro de un núcleo es
decir cómo se dobla como no es lo mismo que bueno sabemos que es una doble hélice que es una doble hélice que se
enrolla sobre sí misma pero luego las estructuras de orden superior es
decir aquellas que tienen un grado de complejidad mayor
Son dos metros de molécula que se empaquetan en
100 nanómetros entonces que es o que tienen el núcleo de diametro
como se dobla esa molécula dentro del núcleo es se está viendo que es algo muy
una pregunta muy relevante y que es realmente
una cuestión que pues determina muchos
aspectos de el desarrollo del embrión o por ejemplo a un tema como la
polidactilia no a la polidactilia se ha visto que a depende hay una zona del
genoma que determina pues de cuántos dedos tenemos en la mano si esa zona del genoma se empaqueta diferente es decir
se dobla sobre sí misma de una forma aberrante se generan fenómenos como la
polidactilia y pues ya se está viendo que hay muchas pues muchas enfermedades
muchos aspectos de la biología que dependen específicamente de cómo se enrolla el dna dentro del núcleo y bueno
pues si es una área de estudio que mi laboratorio pues está explorando no si es un poco
sí porque una de las áreas más activas es estas comparaciones que se hacen entre distintas especies y estas estas
zonas volviendo un poco a las técnicas que utilizamos de una manera muy muy sencilla allí e intentando ya llegar un poco más
a un detalle como digo sin abandonarla a la parte divulgativa conocemos por ejemplo marcas específicas que hacen
cosas concretas es decir sabemos que una proteína de las que se pegan al adn de las que normalmente están pegadas al adn
dándoles parte de esta estructura primaria que yo tiene para que se organice pues hay por ejemplo dos o tres
modificaciones en esa proteína y esas modificaciones más o menos hasta donde sabemos siempre hacen lo mismo ejemplo
activar un gen cuando digo activar es hacer que algo ahora que te produce es y
hay otras que al contrario cuando están esas marcas que a lo mejor es la misma proteína pero es otra marca diferente
como si tuviera dos tapones uno en un sitio y en otro de distintos colores hace pues que no se nos expresen y
estudiando este tipo de marcas comparando unas especies con otras somos capaces de identificar regiones por
ejemplo que son importantes que están conservadas y es una de las cosas con las que se
estudian cuco cómo se como se hace en estos estudios es decir de una manera
sencilla qué tipo de técnicas utilizadas por ejemplo alejandra para saber de detectar por claro nosotros aquí vemos
el tapón de color verde pero como en un laboratorio tú puedes decir pues veo esta marca si antes de hacer un
comentario con respecto con lo que dijiste que es súper relevante nosotros
pensamos que somos especiales pero la verdad es que los mecanismos moleculares que ocurren en nosotros o en una mosca o
en una hormiga son exactamente los mismos el adn
en la bacteria en el mismo los componentes son los mismos lo único que
cambia es la organización entonces hay mucha homología a nivel
molecular en los procesos que ocurren tanto en humanos como en especies como
ratón o qué sé yo mosca y debido a eso podemos usar esos modelos
para investigar hay gente que usa gusanos hay gente que usa levadura hay
gente que usa células humanas y finalmente llegamos a hay variaciones
pero finalmente los mecanismos que ocurren en las distintas especies son los mismos ahora como estudiamos
infinidad de técnicas en mi laboratorio somos bastantes bioquímicos lo que nos gusta es purificar esto o sea a partir
de una gran cantidad de células por ejemplo y llegar a esta proteína y
caracterizar esta proteína investigar cómo funciona en el tubo ensayo si yo le
pongo más temperaturas y le pongo en más azúcar etcétera qué pasa
y una vez que uno entiende esos mecanismos muy básicos puede ir a empezar a variar los a modificar lo
bueno qué pasa si yo le cambio en la la secuencia en desde que tenga este
tubo así que cortar lo que pasa con esa proteína porque eso también nos va a ayudar a entender cómo funciona y quizás
llegar a entender una enfermedad no porque cambia utilizamos entonces métodos de
purificación en este momento debido a toda la tecnología que existe ya hay por
ejemplo secuenciación masiva lo que decías tú recién de la secuenciación del
genoma humano fue además de todo el conocimiento que nos trajo fue una un
abrir y una ayuda técnica pero impresionante y en este momento ya
podemos secuenciar el genoma completo en es barato lo podemos hacer en cualquier
momento podemos secuenciar infinidad de especies en el mismo
momento entonces las técnicas han avanzado muchísimo y ya no estudiamos solamente una
en un momento cuando yo partí mi estudio investigamos esta región pero en este
momento ya se investiga el genoma completo de las células y al mismo tiempo se estudia estas marcas a nivel
de todas las células ya no solamente en una región chiquitita sino que a nivel
general y eso también a seguir es favorecido por técnicas que parten
como investigación básica y después a una investigación a un investigador se le ocurre que se puede aplicar en otra
cosa y entonces así se van generando conocimientos nuevo y también técnicas nuevas que ayudan muchísimo no puedo
entrar en detalle porque no tienen ningún sentido general
minutillos su minuciosa y extraordinaria los que somos un poco más perezosos hacemos otras cosas
utilizamos técnicas que te permiten ver estas marcas sin necesidad de tener que purificarlo porque de verdad es un
trabajo extraordinario de tener que purifica las cosas entonces puedes utilizar una proteína que reconoce la
tapita verde y esa le pega es una cosa que es fluorescente y entonces puedes verlo en la célula en vivo en el
microscopio es la marquita cómo se mueve dentro del núcleo eso hacemos los que hacemos un poco más precios no hay que
aleja nada son técnicas complementarias son y también y en cuanto a la comparación con las especies por ejemplo
nosotros vamos a utilizado en el laboratorio para un problema de regeneración por ejemplo los peces
regeneran la retina y los mamíferos no entonces nosotros queríamos saber qué es
lo que tiene el pez que no tenemos nosotros o qué es lo que tenemos nosotros que molesta y que no debería estar en cuanto a epigenética porque
teníamos pistas de que esto podría ser un problema genético y lo que hicimos fue comparar directamente lo que es lo
que hacen las cosas epigenéticas que ocurren algunas que ocurren en el pp después del daño para que mientras va
regenerando la retina y lo que hace el ratón por ejemplo el mamífero y
buscábamos obviamente diferencias y no similitudes y encontramos algunas cosas que ocurren igual y otras que son
totalmente diferentes cientos nos vamos a poner esas claro en esas actividades epigenéticas que están haciendo si yo la
puedo modular en el laboratorio a lo mejor puede hacer que el ratón regenere la retina
y bueno obviamente hay muchas utilizas el conocimiento decía alejandro muy bien
que la ciencia la hacemos todos en conjunto con muchos laboratorios trabajando utilizas la información que
publica otra gente otros laboratorios para crecer tu propia investigación y
hacer una escalera que va conduciendo no sé si en este sentido otra cosa importante que también hemos oído y
muchas veces le ponemos apellidos a la ciencia ciencia básica ciencia aplicada y a veces por supuesto que queremos
solucionar los problemas y queremos hacerlo lo antes posible pero quizás sea equivocado centrarnos en enfocar la
ciencia en solucionar un problema la ciencia es conocimiento y el conocimiento siempre es importante y siempre va a dar un fruto mañana o
dentro de 20 años en este sentido por ejemplo vuestras investigaciones que pueden parecer que son muy básicas o que
no llevan a ningún sitio o la pregunta que a todos los científicos nos suelen hacer y eso que tú haces para qué sirve sirve para aprender y aprender siempre
está bien y por supuesto aprender cosas de detalle de los que estamos hablando pues pues mucho más querías fabiana
mencionar porque el tema de los anticuerpos a mí me parece importante porque a lo mejor mucha gente ha oído
hablar de anticuerpos como algo de nuestra defensa pero es una de las herramientas más fundamentales que usamos todos los científicos en el
laboratorio a estudiar las marcas epigenéticas por ejemplo si hay una marca amarilla
indicar que un gen se va a activar hay anticuerpos específicos para la marca amarilla la marca roja la marca verde
entonces se se incuban se ponen en distintos ambientes y
después se incuban con esos anticuerpos se precipita se purifica y se identifica que gen está o qué sector del adn está
marcado con esa marca que vamos a saber que se va a activar o se va a inhibir entonces este identificando esas marcas
con anticuerpos específicos de las marcas identificamos qué marcas hay identificamos que hay distintas marcas
de acuerdo en qué en qué ambiente esté esa célula si el tema de los anticuerpos como herramientas de investigaciones es
fascinante y es un ejemplo más de cómo algo que la naturaleza digamos ha producido para un fin que es defendernos
de las infecciones porque recuerdan que un anticuerpo es una molécula es una
molécula que reconoce a otra básicamente entonces normalmente lo que lo que hacen es reconocer por ejemplo
bacterias virus que nos infectan para eliminarlos esa es su función natural si nosotros ahora desarrollamos anticuerpos
contra el tapón amarillo pues somos capaces de las células en el tubo de ensayo usar
estos anticuerpos como una herramienta que nos permite saber y identificar dónde están estos anticuerpos otro tema
que está muy dado muy de moda ahora a lo mejor han oído es el tema de la edición genómica el cris per es una técnica que
lo mismo es una tecnología un ejemplo que unas bacterias desarrollaron para defenderse y que nosotros ahora hemos
utilizado para precisamente cambiar la secuencia del adn de una manera muy
mucho más sencilla de lo que de lo que ocurría hasta hasta ahora
a mí esto me parece tremendamente relevante porque de nuevo con la idea de
que nosotros tenemos que hacer investigación que sea aplicada o que tenga la investigación no necesariamente
nos va a llevar a eso o sea puede que estemos pensando de una manera que no nos va a llevar a ninguna a ninguna parte sin embargo investigaciones como
lo que están mencionando con jose de crisis percas que en la edición higiénica que se está haciendo ahora fueron investigadoras mujeres que
estaban interesadas en invertir en entender cómo la bacteria se defiende o
sea si yo hubiese dicho eso no sé 20 años atrás quizá había medir tubería ti
qué te importa o sea para que no pero sin embargo ese conocimiento está
y bueno después alguien se le ocurrió a espero esto lo podemos utilizar y yo
creo que va a ser una de las revoluciones medicina porque es probable que podamos después editar nuestro genoma no ahora en un tiempo más pero es
una cosa que lo más probable va a pasar y va a revolucionar la medicina entonces uno no sabe por donde la aplicación del
conocimiento va a ocurrir y entonces enfocar la investigación en solamente
aplicación muchos gobiernos lamentablemente en mi país ocurre muchos políticos quieren ver efectos o
resultados inmediatos y la ciencia no es así no es así muy lejos puede que hay un
descubrimiento que se hizo veinte años atrás y que la persona que lo hizo nunca se le ocurrió pero a la persona que lo
leyó a mira esto cómo publicar estas cosas y ahí se genera la aplicación pero
uno no hace una investigación basada en aplicación yo creo que eso es un error que mucha gente tiene y por ahí no va o
sea no podemos no existe la ciencia aplicada no existe es la generación de
conocimiento y en algún momento eso va a llevar aplicación es algo que uno tiene que entender y que
darle los tiempos a los científicos para que investiguen no son los mismos tiempos los políticos que quieren
reelección y entonces quieren resultados de ciencia tiene otros tiempos que son
hermosos no porque nos da tiempo para investigar pero pero no es la
investigación es generación de conocimiento con la potencialidad de
utilizarla
ue está dentro de todas nuestras células es lo que determina pues bueno como somos como son nuestras
características morfológicas cómo son nuestras características fisiológicas muchas veces también qué tipo de
enfermedades vamos a padecer hasta hace poco pensábamos que aquí estaba todo aquí es cuando digo aquí en esta
molécula que normalmente tiene una estructura enrollada como una doble hélice pero si la desarrollamos vemos
que es una secuencia si ven a ustedes aquí una secuencia de componentes
químicos cuatro diferentes que van formando una lista y cada uno de nuestros genes cada uno de
nuestras información genética está compuesta por una combinación diferente de todas estas letras para hacer un
paralelismo aquí están dibujado con colores pues el hecho de que una persona tenga aquí amarillo azul amarillo rojo
bueno pues esa secuencia que son nuestros genes determina como somos y como digo hasta hace poco pensábamos que
ahí estaba todo ahora hemos descubierto que hay algo más y hay algo más
significa que a esta molécula de de ácido nucleico que forman nuestros genes del adn se pegan proteínas son otras
moléculas que son las que hacen las funciones de nuestra en nuestras células y estas moléculas estas proteínas se
pegan y hacen que el adn transmita esa información la información que está aquí contenida está secuencia tiene que
transmitirse a otras moléculas y estas proteínas que se pegan aquí manejan como
función como como es como ocurre esto pues entonces estas proteínas a su vez hay muchas de ellas y pueden tener una
serie de etiquetas por ejemplo aquí hay una que tiene una etiqueta verde una etiqueta amarilla y una
que te arroja para simplificar hablo en colores ustedes imaginen que lo que estamos hablando aquí son de modificaciones químicas bueno pues esto
es la epigenética epigenética lo que está por encima de la genética la genética es la secuencia de nuestros
genes la epigenética es lo que se pone sobre ellos regulando los y de esto es lo que vamos a hablar hoy porque qué
tipo de etiquetas tienen estas proteínas qué tipo de etiquetas a veces tiene también el propio adn va a hacer que se
comporte de una manera de otra y que algunos genes se activen funcionen otros
genes no funcionen correctamente y sabiendo cómo se controla cómo estas
etiquetas se colocan dentro de las moléculas podemos entender pues muchas cosas de cómo funciona la salud porque
quizás es lo que más nos importa pero muchas otras cosas por ejemplo también las plantas los animales tienen
epigenética y controlando estos mecanismos pues podemos entender e
incluso llegar a manipular en nuestro beneficio las características que tienen
entonces hecha esta breve introducción para ponernos en contexto voy a preguntarles a nuestras invitadas
es como se descubrió cuando se empezó a
entender que este tipo de modificaciones existían y de qué manera podemos
estudiarlas alejandra por ejemplo cuando surgió digamos este conocimiento cuando nos dan
nos dimos cuenta de que había estas marcas moleculares yo creo que un hito
importante en este área aunque todavía no se mencionaba como ty genética fueron unos estudios en los años 50 donde por
un premio nobel actualmente john gurdon y kim que ellos estaba la idea de que
las células tenemos distintas células nuestro cuerpo tenemos neuronas tenemos linfocitos que
son los que nos ayudan a responder frente infecciones tenemos células hepáticas que están en el hígado
todas estas células tienen la misma información genética el mismo adn
sin embargo son tremendamente diferentes y en los 50 se pensaba que la
diferenciación celular o sea que nosotros tengamos estas distintas células se debía a que la célula perdía
información genética entonces una célula muscular iba a tener
información genética distinta a una célula de en la neurona porque perdía
información y eso le daba la diferencia pero en ese año entonces se hizo el
primer planeamiento el primer ciclo no una rana desde se sacó desde desde una
célula que ya pertenecía a una rana adulta y se pudo hacer una nueva rana lo
que significa la importancia de ese experimento fue que se demostró que esa célula
que estaba diferenciada en una rana adulta contenía toda la información de
una que podía generar una nueva rana por lo tanto no había pérdida de información entonces cuál es la siguiente idea que
una célula neuronal una célula muscular tiene la
misma información genética pero expresa distintos genes y eso se regula a nivel
genético entonces para mí ese es un experimento que fue tremendamente importante para definir que hay algo más
en el adn que nos permite entonces tener distinto información en distintas células
he mencionado brevemente no sé si alguna puede contribuir a esto que realmente estas modificaciones pueden estar tanto
en la propia secuencia del adn como en las proteínas mónica por ejemplo puedes comentar un
poquito bueno yo quisiera añadir un poco a esta a esta imagen que tú nos has dado
que estas modificaciones estas marcas que se ponen en el adn o en las
proteínas asociadas lo que hacen es que cambian la arquitectura del dna vale
entonces digamos la imagen muy sencilla de entender es que el dna está formado
por ladrillos cuatro ladrillos que son idénticos y que se repiten en un orden determinado
y una serie de grupos químicos que se repiten en un orden determinado uno con unos ladrillos puede hacer a un
apartamentito a las afueras de madrid o el palacio de linares estamos de acuerdo y esto es lo
que hacen estas marcas estas marcas se fijen en la epigenética se refiere justamente a esto a la arquitectura del
dna cómo está organizado así lo que tienes en una zona determinada es el apartamento a las afueras de madrid o es
el palacio de linares y tienes una neurona funcionando a todo lo que da entonces
esto es importante porque durante mucho tiempo nosotros estuvimos
estudiando nosotros me refiero a los biólogos moleculares con mucho detalle
justamente como de mención a alejandra cómo se expresan los genes como hace que hacer que un gen se produzca cómo se
produce la proteína y era como si viéramos está estudiando durante mucho tiempo una parte muy pequeña de algo y
de repente alguien hubiera quitado una cortina y hubiera visto que hay toda una historia detrás inmensa que no
considerábamos y que entonces ahora tenemos que considerar porque la imagen es diferente no es lo mismo estar
estudiando una parte de un dedo de una estatua y luego quitar la cortina y ver que lo que tienes es el david de miguel
ángel por ejemplo esto es lo que ha hecho la epigenética entonces estos son estos son los cambios que
que nosotros tenemos que estudiar ahora sobre todo lo que estudiábamos antes toda esta nueva visión esta parte del
iceberg que está por debajo del agua esto es lo que estamos estudiando y esto es lo que está afectando absolutamente a
cómo vemos las enfermedades cómo entendemos el funcionamiento de las células etc
porque lorena por ejemplo estos cambios que estamos mencionando que son muy muy
variados por una parte es decir hay muchas de estas etiquetas muchas marcas pero además son muy dinámicos es cierto
es decir si ciertamente yo creo que una de las características más importantes
que realmente realzan la relevancia de lo que es la epigenética es precisamente
eso lo que es un mecanismo que tiene gran plasticidad y gran dinamismo y es
reversible porque lo que es el genoma que nos enseñaban nos enseñaba antes con jose con esta molécula el genoma es el
mismo todo el tiempo en todas las células entonces es una molécula que o sea en sí la composición no varía no
acerca de una mutación en teoría no debería de ocurrir aunque ocurre pero no es algo que sea adecuado
lo que posibilita que el dna funcione y se generen los procesos que el propio de
neal debe llevar a cabo son estos mecanismos epigenéticos porque ellos son los que tienen movilidad plasticidad
cambian a dependiendo del tipo celular dependiendo de los estímulos los
estímulos ambientales son los que permiten que el dna ejerza
su función adecuada en cada momento en cada tipo de celular y en respuesta a cada situación ambiental en el en la
cual se encuentra la célula y bueno pues yo creo que sí que este es quizás la
característica que yo encuentro más atrayente para para dedicarme precisamente al estudio de la tijera
ética sí creo que esto que menciona a lorena es es muy importante el tema de las
mutaciones y el tema de la plasticidad el epigenética por supuesto que la secuencia del adn puede puede cambiar
puede haber un error nosotros estamos preparados para que este modelo sea la doble hélice está pensada precisamente
para que se copie a sí misma cada una de las dos cadenas se separan y cada una
sirve de molde para que la otra cadena como si fuera un espejo se se copie sobre ella claro eso es un proceso
mecánico bueno mecánico físico y puede haber errores las células están
preparadas para corregir esos errores pero a veces no pueden con ello entonces hay factores que son capaces de
introducir mutaciones en este adn en la secuencia inicial como comentaba lorena pero normalmente no ocurren porque
estamos preparados para ello y si ocurren pues pueden dar lugar a enfermedades hay factores como puede ser por ejemplo la luz ultravioleta del sol
que puede dañar el adn y por eso tenemos que protegernos porque esas mutaciones se quedan fijadas en el adn una vez que
ya han sucedido y se transmiten de una célula a la siguiente en cambio todo este dinamismo de la epigenética todo
de las etiquetas moleculares también está muy influido por cosas por factores
ambientales que hasta ahora no éramos muy conscientes y que ahora estamos aprendiendo que no solamente por ejemplo
en este caso pues la luz en sí misma sino que la alimentación la polución la
contaminación hay muchos factores externos que pueden tener un impacto y esto creo que es una de las cosas importantes que hemos aprendido verdad
en fabiana sí porque en estas marcas que determinan si un determinado gen se va a expresar o
no este influye el ambiente el ambiente hace cambia estas marcas y entonces hay
marcas que dicen que un determinado gen se va a expresar o que un determinado gen no se va a expresar y entonces
nosotros lo que hacemos es estudiar esas marcas para saber este si si un gen determinado se va a expresar o no en las
plantas por ejemplo que es un área donde yo estudio se ha visto por ejemplo de por qué las plantas no florecen en el
invierno y se ha visto que está implicada la epigenética en eso se ayuna hay una proteína que se expresa en
determinado momento durante el invierno y hace que no se desencadene la floración y cuando cambia el ambiente
cambia la temperatura cambian determinadas condiciones o las condiciones de luz éste se produce una
marca epigenética que hace que una proteína se exprese y entonces la planta florezca y lo maravilloso que se ha
visto también de estas marcas es que por ejemplo en las plantas se heredan o sea es una memoria que si una determinada
planta por ejemplo se vivió en un determinado ambiente y tiene determinadas condiciones
de generaciones de las plantas esas marcas se van manteniendo o sea no sólo se van manteniendo la secuencia del adn
sino que por un tiempo hasta que se marque se desmarquen esas marcas también se heredan entonces es esto de un área
que nos ha cambiado un poco este el paradigma de cómo estudiar algunas algunos aspectos cómo abordar algunos
estudios en bioquímica y en biología molecular que nos ha impactado a todos los científicos pero sí que me gustaría
añadir qué esto qué ocurre en las plantas ocurre también en los humanos y qué es una de
las cosas que justamente la epigenética incide hablando de la pregunta que nos
hacemos hoy de cómo incide sobre nuestra salud lo que hacemos nosotros hoy puede afectar a nuestros nietos y por ejemplo
y esto ha habido experimentos hechos en gente que pasó hambre durante la segunda guerra mundial la famosa hambruna en
holanda hay gente que pasa mucha hambre por el racionamiento de la alimentación los nietos de esta gente hoy en día
podemos seguir las marcas epigenéticas que causó aquella aquella condición y los nietos todavía tienen tienen marcas
epigenéticas en su adn o sea que su adn funciona de una manera particular eso es histórico así fueron los primeros
experimentos que se hicieron sobre estas era de habilidad en el humano y esto implica que hoy en día lo que nosotros
hacemos lo que bebemos lo que comemos el atasco de la m-30 en vivir en la ciudad de méxico en fin estas cosas
influyen sobre mí epigenoma el pez ya no ma de nuestros descendientes también y
es una una cosa más de las que tenemos que tener en cuenta que antes no
conocíamos esto que mencionas mónica es muy interesante porque ciertamente hasta hasta hace poco y yo creo que hoy en día
y controversia también porque claro eso significa que modificaciones de nuestro ambiente son capaces de pasar a
generaciones posteriores y eso digamos que era una va en contra del dogma de la
de la herencia genética pero hoy en día efectivamente hay experimentos que demuestran que estas marcas epigenéticas
algunas la mayoría son muy dinámicas son lo que llamamos reversibles son de quita
y pon pero hay otras que son tan estables que incluso pueden pasar las generaciones es cosas y verdad hay experimentos el que has mencionado y
otros en el laboratorio en el que esto se ha demostrado en el en el yo conozco algunos en particular en el área de la
psiquiatría en la que justamente hay una idea de que digamos estas marcas
epigenéticas al principio me digan por ejemplo en el mundo de las adicciones
las adicciones causan cambios en el epigenoma de la gente son de estas marcas epigenéticas de la persona adicta
y estas marcas pueden ser reversibles durante un tiempo pero si se mantienen y se mantienen al final se hacen estables
y esas marcas epigenéticas se pueden pasar a la progenie incluso la progenie
la puede pasar a la siguiente progenie efectivamente esto ya está comprobado está comprobado que tenemos un nivel
adicional ya no es sólo para nosotros sino para nuestros hijos nietos etc quería que me comentara si alguna en
algún caso concreto de enfermedades por ejemplo en humanos que tengan una base
genética y que esté muy muy claro muy estudiado alguien tiene a lo mejor incluso trabajáis directamente en vuestros laboratorios en este campo
bueno yo creo que es un campo que el campo de la ficción ética es un campo relativamente nuevo que
pues no permite o sea es muy complejo porque hay muchos factores que
influencian hay muchas mucha maquinaria molecular implicada muchas proteínas distintas muchas integraciones redes
genéticas que los coordinan y por tanto el entenderlo pues todavía estamos en en
ello en lo que son lo que se denominan enfermedades complejas como por ejemplo
el tema de la obesidad o el desarrollo de diabetes a enfermedades como
enfermedades neuropsiquiátricas que tienen muchos factores que convergen
para que se dé la enfermedad pues tiene un componente genético muy fuerte pero
es de momento nos está resultando muy difícil discernir digamos encontrar el
elixir hola e la diana molecular que nos va a permitir
resolver estos problemas pero algunos avances se hacen por ejemplo en cáncer algunos tipos de cáncer
lo que serían los tratamientos para curarlos pues hay algunos tipos de tratamiento tratamientos epigenéticos
que permiten son químicos que modulan
algunas de las de alguna de la máquina molecular implicada en la
en el control del epigenoma y pues han probado efectivos para el tratamiento de
ciertos cánceres de ciertos cánceres muy concretos y bueno yo creo que ahí el campo es donde quizás esté más avanzado
en mi opinión no sé si ustedes conozcan bien yo creo que lo que pasa es que en
la mayoría si no todas las enfermedades finalmente se reducen a que hay un
cambio en en la expresión de algunos factores
y eso está regulado por la parte genética y por la parte de genética
yo diría que los años noventas la investigación estuvo centrada en
identificar osea purificar desde desde una célula que se crece en cultivo
llegar hasta la proteína que producía la función y el hilo como dice juan jose es
buscar qué proteína ponía esta tapita verde acá
y entonces fueron muchísimos años décadas donde se hizo eso muchísimos
laboratorios la ciencia se hace en conjunto y nosotros tenemos mucho mucha comunicación entre los científicos
publicamos nuestros datos entonces ustedes pueden buscarlo es disponible
una vez que se identificaron estas proteínas que son tremendamente relevantes
los científicos nos empezamos a dar cuenta que muchas de esas proteínas estaban montadas en algunas enfermedades
la mayoría de las proteínas que van a producir la tapita verde la tapita amarilla roja están mutadas en muchas
enfermedades son diferentes o funcionan más o funcionan menos o no funcionan
enfermedades como el cáncer son enfermedades multifactoriales no se debe
a una modificación a una a una falla sino que son muchas entonces es difícil
definir que una proteína sea ay hay enfermedades donde si uno puede decir un
factor hemofilia por ejemplo se debe a que el factor 8 no se produce y eso es
porque hay un problema a nivel de adn puede ser o también puede ser un problema a nivel de la regulación de
como ese factor se expresa
y la pregunta se me falta algún ejemplo
concreto y lo que está quedando claro es que los mecanismos son tan complejos son multifactoriales son multifactoriales
hay casos de enfermedades monogénicas que es como nos referimos a ellas que un único gen o una única un único problema
en un factor concreto causan la enfermedad y puede ser una enfermedad gravísima si lo que está modificado lo
que no funciona bien es muy importante pero normalmente las enfermedades sobre todo las enfermedades más prevalentes
las que afectan a la mayoría de la población suelen tener origen es multifactorial es entonces muchas veces
es muy difícil acotar cuál es el problema y estamos hablando a lo mejor simplemente de la secuencia del adn si
ya metemos todo el tema epigenético es complicado pero es que es justamente ahí
es justamente porque hubo un momento que todos
recordarán a veces la ciencia salta del laboratorio a la calle y uno empieza a
ver en los periódicos el intermediario que se habla de genes y que hoy y que en
el gimnasio te hacen el análisis de análisis genómico para ver si vas a poder subir tres escaleras o no en fin y
hubo un momento en el que todos pensábamos que el dna era definitivo o
sea que tú nacieras con un dna y esa era tu destino incluso había una película no sé si recuerdan llamaba gattaca en o que
el de línea fija va a tu destino en la vida y ya no podía ser astronauta si no
tenías un cierto dna y justamente había preguntas en ese
entonces en este concepto por eso se lanzó el proyecto del genoma humano vamos a leer el genoma humano entero vamos a ver todos los ladrillos que
constituyen el genoma humano y lo vamos a entender y vamos a solucionar todo y vamos a entender todas las enfermedades
que no se conocían porque vamos a encontrar todos estos genes que no están funcionando y entonces
se resuelve el genoma humano se lee y diez años después seguimos más o menos exactamente en la misma posición no
entendemos nada ni sabemos nada de estas enfermedades esto es justamente lo más interesante en los laboratorios cuando
un experimento nos sale como uno espera porque entonces te tiene que poner a pensar y entonces en este caso todas
esas preguntas que no se podían resolver porque dos gemelos que tienen exactamente el mismo dna el mismo dna no
son iguales y uno a los veinte años desarrolla esquizofrenia y el otro no porque no hemos conocido más solución a
enfermedades no después de haber leído todo el genoma humano y habernos gastado millones de dólares y porque hemos
avanzado muy poco en el conocimiento pues porque las cosas no iban por ahí no rascó y cómo y cómo era esta historia
mágica de no el ambiente interfiere en el dna porque un gemelo es esquizofrénico nungwi es el ambiente no
pero no teníamos ni la más remota idea de cómo el ambiente intervenía bueno todo eso es la epigenética como el ambiente interfiere como ya en el genoma
no dicta nuestro futuro porque el epigenoma como bien dicen todas mis
compañeras se puede cambiar y si tú puedes afectar tu epigenoma y puedes cambiar el destino de esto y en
las enfermedades pues en particular mucho esfuerzo realizado para encontrar
un gen que fuera responsable de una enfermedad estudiando a las familias el afectado la familia a la abuela la
bisabuela el que viene de la argentina el otro sector mont y no se encontraban los genes porque porque no estaba en los
genes estaba en la epigenética estaba en la arquitectura estaba en estas marcas que hacen que el genoma sea diferente
entonces ahí es justo todo esto es justo donde en la epigenética ha venido a cambiarnos la perspectiva si yo también
quería añadir que en ese misma en esa misma línea que o sea alejándola nos ha
dicho hace un rato que en los años 50 mil o 150 si se comenzaba a hablar de este tipo de conceptos epigenéticas fue
acuñada pues la palabra en concreto por aquella época pero es un campo relativamente nuevo no porque no ha sido
hasta finales de los 90 años 2000 o así que ha empezado a crecer exponencialmente gracias a las nuevas
tecnologías y a las nuevas capacidades que tenemos para entender este tipo de problemas y entonces bueno pues no ha
pasado tanto en un tiempo largo que tiene de historia
la ciencia pues el campo de la epigenética es un campo muy nuevo no entonces pues todavía muchas cosas por
describir y por descubrir y creo que te va a dar muchos más frutos de los que
está dando y todavía hay muchas cosas que que podemos entender y que nos van a ayudar a comprender ciertas enfermedades
de relevancia mundial ahora mismo en esa misma línea
si hay varias cosas mónica que has mencionado que son muy interesantes tenía aquí y de hecho ha notado a sacarle el tema por ejemplo
particularmente de los gemelos que es el paradigma de las personas genéticamente idénticas y es así y muchas veces o el
argumento que se ha utilizado históricamente para defender esto es gemelos a lo mejor que son separados y
luego son iguales en aficiones en comportamental pero tú has mencionado el caso contrario que efectivamente es lo que nos estamos dando cuenta casos en
los que son idénticamente genéticos genéticamente igual pero por la epigenética en este caso han cambiado y
en ese sentido creo que también hay estudios que hablan de una genética de la edad es decir la edad el
envejecimiento o cómo vamos avanzando a lo largo de la vida parece ser que hay marcas concretas identificadas que
caracterizan esto creo que eso es así verdad alguien quiere comentar algo
bueno sí o sabe que es así así es claro que todavía no sabemos exactamente
desgraciadamente así es que tenemos marcas verdad pero no sabemos cómo eliminarlas digamos
por lo menos en mamíferos en plantas cómo está el asunto
el ambiente cambió muchísimo el epigenoma las marcas que se ven en distintos ambientes en plantas que
tienen el mismo adn es osea cambia muchísimo de acuerdo a si la
planta crece por ejemplo con más tiempo de luz con menos tiempo de luz se ven marcas distintas y se expresan genes
distintos y el adn es el mismo siendo a esto que contabas hay un experimento
también clásico con ratones este en los cuales a las madres tienen a los ratones
con el mismo este mismo material genético pero la alimentación de la
madre durante el embarazo influye en que el ratón salga distinto y salga con por ejemplo este exprese una
determinada enfermedad o no y en la alimentación de la madre que le transmite al hijo y salen distintos la
descendencia de acuerdo a ese proceso sea que el ambiente la alimentación donde nos movemos influye muchísimo en
estas marcas influye muchísimo después en lo que se va a expresar quisiera sobre eso es que es muy bonito a la
nutrición y la epigenética porque además se lo van a encontrar en el supermercado cualquiera
las revisiones científicas lo que hacen es tabular poner en una tabla que
componente epigenético se ha descrito por ejemplo que se yo en el caldo de pollo en el ajo el nose que edita yo
cuando hago todas mis clases le digo a estudiantes que toda esta epigenética lo que está haciendo es confirmar que la
abuela tenía razón y que la isla de bueno comer el brócoli y que no es muy
bueno tomar mucho azúcar y todo esto explicado desde un punto de vista molecular es parte de lo que ha pasado en el congreso esta bioquímica en la
ciudad que han hablado de etapas y la biología molecular de las tapas y la bioquímica de las tapas bueno pues esto
es exactamente iba con la epigenética y cuando digo que se lo encuentra en el que se va encontrar en la calle es real
porque ya ahí si ustedes buscan en internet epigenética y comida por
ejemplo yo busqué en internet en algún momento para dar una clase y había comida para perros epigenética
epigenética no sé que se llama creo que te dan un perro buenísimo y creo que no
hay todavía para humanos y yo le dije a los estudiantes el primero que me traigo un yogurt epigenético le regaló una botella de tequila ya son ya son mayores
mis estudiantes es donde doctorado todos los de aquí y como locos no encontraron porque es mucho más difícil hacer una
regulación de comida para humanos pero de perro ya la pueden encontrar esto es como la ciencia sal salta del
laboratorio a la calle yo creo que por eso estas sesiones son buenísimas porque de esta manera podemos
conocer y saber que nos está tomando el pelo en la opinión crítica claro que si mónica el comida epigenética mejor no
pero jabón epigenético pues con sabor ya pues hay que empezar a sacar la botella
o sea que entonces claro tenemos tenemos unas marcas epigenéticas de envejecimiento que hemos dicho que bueno
es una cosa que nos gustaría a todos aprender más y controlar verdad pero precisamente también por la epigenética
otro descubrimiento fundamental y aquí es muy relevante lo que contaba alejandra al principio sobre el premio
nobel de de jong cartoon porque compartió el premio nobel con otro investigador en este caso japonés shinya
yamanaka que lo que hizo fue algo no es una palabra que un científico debiera
utilizar pero para que me entiendan coloquialmente algo milagroso y es el tema todo el tema de las células madre
inducidas pluripotentes inducidas no sé si se habrán oído hablar de ello y
básicamente lo que lo que consiguió y lo que se ha conseguido y es uno de los campos más activos hoy en día de trabajo
en los laboratorios es precisamente volver atrás este reloj o estas marcas epigenéticas como ha comentado alejandra
a medida que cuando somos embriones y las células empiezan a diferenciarse se van añadiendo distintas marcas para que
una célula sea de una manera o de otra ahora hemos aprendido métodos mecanismos para borrar
esas marcas y volver hacia atrás de forma que una célula adulta sea capaz de
tener volver hacia un estado un estado pluripotente esto significa que esa
célula ahora la podemos convertir otra vez en otra célula diferente porque
porque estas marcas que se han ido añadiendo más o menos conseguimos
borrarlas y ahora utilizando un cóctel diferente les convertimos en otra cosa
veo que mónica no está muy bien es que es justamente mi tema de trabajo vengan las células basales l aún es diferente a
una experta no fue muy importante el edil es del premio nobel de yamanaka por este proceso por el que introducía
cuatro genes en una célula y la convertía en una célula primigenia digamos una célula capaz de dar lugar a
todas las células del organismo me dieron el premio nobel y tropecientosmil laboratorios en el mundo
empezaron a tratar de justamente utilizar esta técnica para tener células una fuente ilimitada ilimitada de
células para poder generar con las células que a mí me hicieran falta que produjeran insulina o células de
neuronas para la enfermedad de parkinson que se yo muchos laboratorios
y de repente se empezaron a dar cuenta de que estas células sí o sea en algunos
casos si hacían lo que tuvo querías que hicieran durante un tiempo y luego dejaban de hacerlo y volvían a su estado
original más o menos y alguien en estado muy poético dijo estas células tienen
memoria memoria epigenética es decir que tú tienes una célula que
tiene una arquitectura tiene una célula que tiene el palacio de linares dentro y tú puedes al revés tienes tu célula con
tu pisito en chamartín y entonces tratas de cambiarlo y tratas de hacer para orinar
y la célula empieza a hacer las cosas empiezan a cambiar empieza a ahorrar marcas empiezan de marcas nuevas pero
tiene la tendencia a volver a ser tu pisito no voy a volver a hacer lo que era eso es lo que se llama memoria
epigenética es que justamente les voy a hablar mañana en el congreso eso entonces muchos investigadores y el
propio yamanaka después de que le dieron el premio nobel dijo bueno sí sí claro esto es lo que llevo descubrió muy
importante pero claro tiene también aquellos sus cursos sus problemas
vamos el mensaje es por supuesto el avance es impresionante y es un poco lo
que tú decías al principio se generan nuevas preguntas qué es esta memoria epigenética si yo puedo de verdad
solucionarse a que la célula ya se olvide de lo que fue y que realmente se cometan hacerlo por impotente habrá un
pasado gigantesco o sea que entonces estamos todavía hemos conseguido saber que estas marcas son
importantes y que borrando podemos volver a un estado primigenio pero no es tan sencillo es mucho más complejo
vamos las conclusiones hay que seguir investigando claro bueno eso por eso siempre eso está claro y si me permiten
agua aquí el inciso de que es una cosa que como científicos como sociedad española de bioquímica y científicos
pero también como ciudadano tenemos que apoyar y este tipo de actividades también son muy importantes para que la
gente comprenda la importancia por todo lo que estamos debatiendo espero que les estemos convenciendo de que es muy importante estudiar la epigenética en
este caso que es el tema que nos ocupa hoy debemos presionar a nuestros mandatarios a nuestros políticos para
que la inversión en el sistema de investigación tanto en nuestro país como en cualquier otro país
pues sea apoyada de la forma adecuada para poder progresar tenía que otra
pequeña nota que me he hecho respecto a lo que comentábamos de esta estructura del adn porque un término que cuando yo
estudiaba y tampoco hace tanto pero bueno ya unos cuantos años solíamos hablar de él o nos hablaban del
adn basura este es un tema interesante todas estas secuencias que les contaba
al principio los genes cuando se empezó a estudiar el genoma humano nos dimos cuenta de que el porcentaje de toda la
cadena de adn que tenía información genética transmisible a una proteína es
decir algo que sabíamos que funcionaba era muy pequeña y que la mayoría del adn
pues no se sabía para qué estaba ahí y dijo no sé nada de basura no sirve para nada este término que es bastante
desafortunado y hoy sabemos que no es así porque precisamente esta arquitectura compleja que estamos
haciendo y muchas de las modificaciones y muchas de la información no para construir el ladrillo sino la
información que dice construye este ladrillo o cuando lo tiene que construir o cuánto lo tiene que construir o
durante cuánto tiempo está en en esta basura que hoy sabemos que no es
así y esto tiene también muchas implicaciones por una parte para las enfermedades porque estos cambios que
estamos hablando pueden ocurrir en zonas del adn que cuando las leemos pues parece que no son nada y tienen muchas
implicaciones evolutivas porque claro así es como la evolución me va trabajando es decir modificando no a lo
mejor la propia información sino la cuando se tienen que expresar los genes
es lo que lo que hace diferentes unas especies de otras esto es esto si queréis comentar algo al
respecto del tri dimensional del genoma pues es una nueva capa regulatoria que
se viene estudiando desde hace relativamente poco que también se considera como una capa
epigenética porque no depende de la secuencia del dna que es cómo se empaqueta el dna dentro de un núcleo es
decir cómo se dobla como no es lo mismo que bueno sabemos que es una doble hélice que es una doble hélice que se
enrolla sobre sí misma pero luego las estructuras de orden superior es
decir aquellas que tienen un grado de complejidad mayor
[Música] bueno tenemos que en cuenta que son dos metros de molécula verdad que se empaquetan en 10 nanómetros que es lo
que mide el núcleo de diámetro perdón un 100 nanómetros entonces
como se dobla esa molécula dentro del núcleo es se está viendo que es algo muy
una pregunta muy relevante y que es realmente
una cuestión que pues determina muchos
aspectos de el desarrollo del embrión o por ejemplo a un tema como la
polidactilia no a la polidactilia se ha visto que a depende hay una zona del
genoma que determina pues de cuántos dedos tenemos en la mano si esa zona del genoma se empaqueta diferente es decir
se dobla sobre sí misma de una forma aberrante se generan fenómenos como la
polidactilia y pues ya se está viendo que hay muchas pues muchas enfermedades
muchos aspectos de la biología que dependen específicamente de cómo se enrolla el dna dentro del núcleo y bueno
pues si es una área de estudio que mi laboratorio pues está explorando no si es un poco
sí porque una de las áreas más activas es estas comparaciones que se hacen entre distintas especies y estas estas
zonas volviendo un poco a las técnicas que utilizamos de una manera muy muy sencilla allí e intentando ya llegar un poco más
a un detalle como digo sin abandonarla a la parte divulgativa conocemos por ejemplo marcas específicas que hacen
cosas concretas es decir sabemos que una proteína de las que se pegan al adn de las que normalmente están pegadas al adn
dándoles parte de esta estructura primaria que yo tiene para que se organice pues hay por ejemplo dos o tres
modificaciones en esa proteína y esas modificaciones más o menos hasta donde sabemos siempre hacen lo mismo ejemplo
activar un gen cuando digo activar es hacer que algo ahora que te produce es y
hay otras que al contrario cuando están esas marcas que a lo mejor es la misma proteína pero es otra marca diferente
como si tuviera dos tapones uno en un sitio y en otro de distintos colores hace pues que no se nos expresen y
estudiando este tipo de marcas comparando unas especies con otras somos capaces de identificar regiones por
ejemplo que son importantes que están conservadas y es una de las cosas con las que se
estudian cuco cómo se como se hace en estos estudios es decir de una manera
sencilla qué tipo de técnicas utilizadas por ejemplo alejandra para saber de detectar por claro nosotros aquí vemos
el tapón de color verde pero como en un laboratorio tú puedes decir pues veo esta marca si antes de hacer un
comentario con respecto con lo que dijiste que es súper relevante nosotros
pensamos que somos especiales pero la verdad es que los mecanismos moleculares que ocurren en nosotros o en una mosca o
en una hormiga son exactamente los mismos el adn
en la bacteria en el mismo los componentes son los mismos lo único que
cambia es la organización entonces hay mucha homología a nivel
molecular en los procesos que ocurren tanto en humanos como en especies como
ratón o qué sé yo mosca y debido a eso podemos usar esos modelos
para investigar hay gente que usa gusanos hay gente que usa levadura hay
gente que usa células humanas y finalmente llegamos a hay variaciones
pero finalmente los mecanismos que ocurren en las distintas especies son los mismos ahora como estudiamos
infinidad de técnicas en mi laboratorio somos bastantes bioquímicos lo que nos gusta es purificar esto o sea a partir
de una gran cantidad de células por ejemplo y llegar a esta proteína y
caracterizar esta proteína investigar cómo funciona en el tubo ensayo si yo le
pongo más temperaturas y le pongo en más azúcar etcétera qué pasa
y una vez que uno entiende esos mecanismos muy básicos puede ir a empezar a variar los a modificar lo
bueno qué pasa si yo le cambio en la la secuencia en desde que tenga este
tubo así que cortar lo que pasa con esa proteína porque eso también nos va a ayudar a entender cómo funciona y quizás
llegar a entender una enfermedad no porque cambia utilizamos entonces métodos de
purificación en este momento debido a toda la tecnología que existe ya hay por
ejemplo secuenciación masiva lo que decías tú recién de la secuenciación del
genoma humano fue además de todo el conocimiento que nos trajo fue una un
abrir y una ayuda técnica pero impresionante y en este momento ya
podemos secuenciar el genoma completo en es barato lo podemos hacer en cualquier
momento podemos secuenciar infinidad de especies en el mismo
momento entonces las técnicas han avanzado muchísimo y ya no estudiamos solamente una
en un momento cuando yo partí mi estudio investigamos esta región pero en este
momento ya se investiga el genoma completo de las células y al mismo tiempo se estudia estas marcas a nivel
de todas las células ya no solamente en una región chiquitita sino que a nivel
general y eso también a seguir es favorecido por técnicas que parten
como investigación básica y después a una investigación a un investigador se le ocurre que se puede aplicar en otra
cosa y entonces así se van generando conocimientos nuevo y también técnicas nuevas que ayudan muchísimo no puedo
entrar en detalle porque no tienen ningún sentido general
minutillos su minuciosa y extraordinaria los que somos un poco más perezosos hacemos otras cosas
utilizamos técnicas que te permiten ver estas marcas sin necesidad de tener que purificarlo porque de verdad es un
trabajo extraordinario de tener que purifica las cosas entonces puedes utilizar una proteína que reconoce la
tapita verde y esa le pega es una cosa que es fluorescente y entonces puedes verlo en la célula en vivo en el
microscopio es la marquita cómo se mueve dentro del núcleo eso hacemos los que hacemos un poco más precios no hay que
aleja nada son técnicas complementarias son y también y en cuanto a la comparación con las especies por ejemplo
nosotros vamos a utilizado en el laboratorio para un problema de regeneración por ejemplo los peces
regeneran la retina y los mamíferos no entonces nosotros queríamos saber qué es
lo que tiene el pez que no tenemos nosotros o qué es lo que tenemos nosotros que molesta y que no debería estar en cuanto a epigenética porque
teníamos pistas de que esto podría ser un problema genético y lo que hicimos fue comparar directamente lo que es lo
que hacen las cosas epigenéticas que ocurren algunas que ocurren en el pp después del daño para que mientras va
regenerando la retina y lo que hace el ratón por ejemplo el mamífero y
buscábamos obviamente diferencias y no similitudes y encontramos algunas cosas que ocurren igual y otras que son
totalmente diferentes cientos nos vamos a poner esas claro en esas actividades epigenéticas que están haciendo si yo la
puedo modular en el laboratorio a lo mejor puede hacer que el ratón regenere la retina
y bueno obviamente hay muchas utilizas el conocimiento decía alejandro muy bien
que la ciencia la hacemos todos en conjunto con muchos laboratorios trabajando utilizas la información que
publica otra gente otros laboratorios para crecer tu propia investigación y
hacer una escalera que va conduciendo no sé si en este sentido otra cosa importante que también hemos oído y
muchas veces le ponemos apellidos a la ciencia ciencia básica ciencia aplicada y a veces por supuesto que queremos
solucionar los problemas y queremos hacerlo lo antes posible pero quizás sea equivocado centrarnos en enfocar la
ciencia en solucionar un problema la ciencia es conocimiento y el conocimiento siempre es importante y siempre va a dar un fruto mañana o
dentro de 20 años en este sentido por ejemplo vuestras investigaciones que pueden parecer que son muy básicas o que
no llevan a ningún sitio o la pregunta que a todos los científicos nos suelen hacer y eso que tú haces para qué sirve sirve para aprender y aprender siempre
está bien y por supuesto aprender cosas de detalle de los que estamos hablando pues pues mucho más querías fabiana
mencionar porque el tema de los anticuerpos a mí me parece importante porque a lo mejor mucha gente ha oído
hablar de anticuerpos como algo de nuestra defensa pero es una de las herramientas más fundamentales que usamos todos los científicos en el
laboratorio a estudiar las marcas epigenéticas por ejemplo si hay una marca amarilla
indicar que un gen se va a activar hay anticuerpos específicos para la marca amarilla la marca roja la marca verde
entonces se se incuban se ponen en distintos ambientes y
después se incuban con esos anticuerpos se precipita se purifica y se identifica que gen está o qué sector del adn está
marcado con esa marca que vamos a saber que se va a activar o se va a inhibir entonces este identificando esas marcas
con anticuerpos específicos de las marcas identificamos qué marcas hay identificamos que hay distintas marcas
de acuerdo en qué en qué ambiente esté esa célula si el tema de los anticuerpos como herramientas de investigaciones es
fascinante y es un ejemplo más de cómo algo que la naturaleza digamos ha producido para un fin que es defendernos
de las infecciones porque recuerdan que un anticuerpo es una molécula es una
molécula que reconoce a otra básicamente entonces normalmente lo que lo que hacen es reconocer por ejemplo
bacterias virus que nos infectan para eliminarlos esa es su función natural si nosotros ahora desarrollamos anticuerpos
contra el tapón amarillo pues somos capaces de las células en el tubo de ensayo usar
estos anticuerpos como una herramienta que nos permite saber y identificar dónde están estos anticuerpos otro tema
que está muy dado muy de moda ahora a lo mejor han oído es el tema de la edición genómica el cris per es una técnica que
lo mismo es una tecnología un ejemplo que unas bacterias desarrollaron para defenderse y que nosotros ahora hemos
utilizado para precisamente cambiar la secuencia del adn de una manera muy
mucho más sencilla de lo que de lo que ocurría hasta hasta ahora
a mí esto me parece tremendamente relevante porque de nuevo con la idea de
que nosotros tenemos que hacer investigación que sea aplicada o que tenga la investigación no necesariamente
nos va a llevar a eso o sea puede que estemos pensando de una manera que no nos va a llevar a ninguna a ninguna parte sin embargo investigaciones como
lo que están mencionando con jose de crisis percas que en la edición higiénica que se está haciendo ahora fueron investigadoras mujeres que
estaban interesadas en invertir en entender cómo la bacteria se defiende o
sea si yo hubiese dicho eso no sé 20 años atrás quizá había medir tubería ti
qué te importa o sea para que no pero sin embargo ese conocimiento está
y bueno después alguien se le ocurrió a espero esto lo podemos utilizar y yo
creo que va a ser una de las revoluciones medicina porque es probable que podamos después editar nuestro genoma no ahora en un tiempo más pero es
una cosa que lo más probable va a pasar y va a revolucionar la medicina entonces uno no sabe por donde la aplicación del
conocimiento va a ocurrir y entonces enfocar la investigación en solamente
aplicación muchos gobiernos lamentablemente en mi país ocurre muchos políticos quieren ver efectos o
resultados inmediatos y la ciencia no es así no es así muy lejos puede que hay un
descubrimiento que se hizo veinte años atrás y que la persona que lo hizo nunca se le ocurrió pero a la persona que lo
leyó a mira esto cómo publicar estas cosas y ahí se genera la aplicación pero
uno no hace una investigación basada en aplicación yo creo que eso es un error que mucha gente tiene y por ahí no va o
sea no podemos no existe la ciencia aplicada no existe es la generación de
conocimiento y en algún momento eso va a llevar aplicación es algo que uno tiene que entender y que
darle los tiempos a los científicos para que investiguen no son los mismos tiempos los políticos que quieren
reelección y entonces quieren resultados de ciencia tiene otros tiempos que son
hermosos no porque nos da tiempo para investigar pero no es la
investigación es generación de conocimiento con la potencialidad de
utilizarla
…
quiero contarte la historia de Anne y Marie, dos gemelas idénticas nacidas
en el mismo lugar, criadas en la misma casa y, como gemelas que eran, increíblemente
parecidas.
Sin embargo, esas similitudes no duraron para siempre:
A los 30 años, Marie fue diagnosticada con trastorno bipolar, mientras que Anne siguió
manteniendo un buen estado de salud mental.
No obstante, a los 40 años Anne fue diagnosticada de enfermedad cardíaca grave, mientras que
Marie se mantuvo en buena forma física hasta muy avanzada edad.
No solo eso sino que, a medida que pasaban los años, las hermanas gemelas se iban pareciendo
cada vez menos.
Este ejemplo concreto es ficticio, pero como este, existen muchísimos casos reales que
muestran cómo gemelos monocigóticos, es decir, gemelos que surgieron del mismo óvulo
fecundado y que por tanto comparten exactamente la misma información genética, han sufrido
enfermedades muy diferentes y han ido sumando diferencias a medida que pasaban los años.
PERO si nuestra información genética realmente dicta lo que somos y cómo funcionamos, ¿Cómo
pueden ser tan diferentes si comparten el mismo ADN?
Hoy en la Hiperactina, hablamos de EPIGENÉTICA.
Durante mucho tiempo y en varios vídeos del canal hemos dicho que el ADN de nuestras células
contiene la información genética, que define quiénes somos, cuáles son nuestras características.
Y esto es en cierto modo verdad: es el motivo por el que una alteración genética, por
ejemplo un cambio en la secuencia de información que conforma el ADN puede dar lugar a enfermedades
tan conocidas como el cáncer, la fibrosis quística o el daltonismo.
Sin embargo, aquello que dicta lo que eres, cómo funciona tu cuerpo e incluso las enfermedades
que puedes desarrollar en un momento concreto no solo viene dado por tu ADN, sino por la
forma en la que este se lee.
A ver, dentro de lo que es la larga secuencia del ADN, los «genes» son fragmentos dentro
de esa secuencia que contienen la información para sintetizar las proteínas, esas moléculas
que necesita la célula para funcionar.
Concretamente, cada gen tiene la información para sintetizar un tipo de proteína. pues
cuando un gen es “leído” y da lugar a su proteína, decimos que ese gen se está
«expresando».
Es decir, el gen está activo, está teniendo un efecto en la célula.
Y la gracia de esto es que esta expresión de genes va variando en función del momento,
es decir, la célula activa y desactiva los genes según lo que necesite en cada momento,
lo cual es súper importante porque le permite adaptarse a los cambios constantes en su entorno.
Por ejemplo, imagínate que estás cocinando y te quemas con la sartén.
En otras palabras (más biomédicas), se ha producido un daño en un tejido en el que
han muerto células que deberán ser reemplazadas.
Para reparar ese tejido dañado, las células de alrededor de la herida deberán dividirse
para dar lugar a nuevas células, es decir, tendrán que activar los genes involucrados
en la división celular.
O sea, que la célula necesita encender y apagar genes según le convenga, pero ¿Cómo
hace esto?
¿Cómo se consigue activar y desactivar un gen a tu antojo?
Pues precisamente a través de la llamada epigenética.
Epigenética significa “por encima de la genética”, y básicamente consiste en añadir
o quitar pequeñas moléculas químicas en zonas concretas capaces de activar o desactivar
genes.
Es decir, son marcas químicas que cambian la forma en la que se lee el ADN, pero sin
alterar su secuencia genética, que en el fondo sigue siendo la misma.
Por ejemplo, imagina la frase “No está mal la homeopatía”.
Si te digo esto, seguramente entiendas que no tengo una mala opinión sobre la homeopatía,
mientras que si le añado algunos signos de puntuación… el significado de la frase
cambia totalmente.
Haciendo un símil con el ADN, podríamos decir que el ADN vendrían a ser las palabras
de un texto, y la epigenética serían los signos de puntuación que harían que se leyese
de una forma u otra.
Vale, todo esto suena muy abstracto, pero yendo a la chicha, a la biomedicina, ¿Qué
significa exactamente?
¿Qué le hace la epigenética a nuestros genes para cambiar la forma en la que se expresan?
Pues prepárate para ver una vez más por qué nuestro cuerpo es increíblemente complejo…
y fascinante.
Como hemos dicho, las modificaciones epigenéticas consisten en añadir pequeñas moléculas
químicas en sitios muy bien pensados del ADN.
En este vídeo, vamos a ver concretamente dos formas de cambiar la expresión de un
gen: alterando directamente el ADN o alterando las proteínas que empaquetan el ADN.
Empecemos por esta última.
Una de las cosas que más me fascinan del cuerpo humano es la burrísima longitud que
tiene el ADN de nuestras células: dentro de cada una de nuestras microscópicas células
se guarda un total de 2 metros de ADN.
¿Cómo salen las cuentas?
Pues eso es así porque el ADN no está todo desparramado por la célula, sino que se encuentra
ultra empaquetado de forma muy bien estructurada y diseñada gracias a unas proteínas llamadas
histonas, sobre las que se enrolla el ADN para empaquetarse.
Si entendemos el ADN como una secuencia de información genética que la célula necesita
“leer”, es fácil imaginar que si ese ADN está muy empaquetado, este será ilegible.
Básicamente porque las proteínas que se encargan de “leer” el ADN no podrán acceder a él correctamente.
Por tanto, los genes que se encuentren en zonas del ADN muy empaquetadas estarán inaccesibles y por tanto esos genes estarán “apagados”, no se van a expresar.
¿Qué debemos hacer si queremos activar esos genes?
Pues siguiendo la misma lógica: desenrollar el ADN.
Cuando las histonas desenrollan el ADN, esa información vuelve a estar accesible y sus genes pueden e: expresarse están “activos”… papi.
¿Y cómo controla una célula que las histonas desenrollen o enrollen los fragmentos de ADN que le interesan? Pues a través de la epigenética.
La célula añadirá pequeñas moléculas químicas a las histonas que cambiarán su estructura y por tanto el empaquetamiento del ADN.
Aunque existen otros ejemplos, una de las moléculas químicas que pueden hacer esto es el llamado grupo acetilo: cuando se añade un grupo acetilo a una histona, esta cambia su estructura de forma que el empaquetamiento se relaja, y el ADN pasa a estar accesible para leerse.
Pero la epigenética es reversible: también podemos hacer lo contrario, quitar ese grupo
acetilo, de forma que la histona, vuelve a cambiar su estructura, compacta de nuevo el
ADN y este ya no se puede leer.
Y este juego tan tonto de empaquetar y desempaquetar el ADN es un ejemplo de cómo las células
pueden tener algunos genes activos mientras que otros están silenciados.
Algo que permitió, entre otras cosas, que nuestras células se especializasen en funciones
muy concretas, dando lugar a distintos tipos celulares.
Aquí viene un dato mindblowing de la biomedicina: todas las células de tu cuerpo tienen el
mismo ADN, los mismos genes, ya sea una célula hepática, una neurona o una célula de tu
retina.
WTF, ¿Cómo es posible si son tan diferentes?
La cosa es que gracias a la epigenética que permite modificar la estructura de las histonas
y cómo se empaqueta y se lee ese ADN, cada célula tendrá activos únicamente los genes
que le interesen: por ejemplo, una célula del hígado expresará genes que le permitan
metabolizar ciertos fármacos, pero no le interesarán los genes que nos permitan ver
con nuestros ojos, mientras que en una célula de la retina pasará lo contrario.
En resumen: la modificación de las histonas es una forma útil de cambiar la expresión
de los genes a nuestro antojo, pero existe una forma de epigenética más directa, que
en lugar de afectar a las proteínas que empaquetan el ADN, afecta directamente al ADN.
La forma más sencilla de epigenética y también la más estudiada, es la llamada metilación
del ADN.
Se llama así porque consiste en añadir una molécula química llamada “grupo metilo”
a las bases nitrogenadas del ADN, concretamente a las citosinas, para que veáis hasta qué
punto es todo ultra específico.
Este grupo metilo, ahora unido a la molécula de ADN, va a impedir que se le adhieran las
proteínas necesarias para leer el gen en concreto, con lo que este quedará inactivado.
Sería algo así como si tuviésemos un libro (que representaría todo el ADN, toda la información
genética) y esta página fuera un gen.
Si le pusiéramos una pegatina enorme, estaríamos impidiendo que la persona encargada de leer
este libro pudiese hacerlo, bloqueando la lectura de esa página.
Por suerte, como hemos comentado, la epigenética es un proceso reversible: igual que podemos
añadir el grupo metilo que “bloquea” el gen, también existe el proceso contrario,
la desmetilación, en la cual se retira ese grupo metilo y el gen se activa.
Lo mismo pasaba con las histonas que hemos visto: añadir un grupo acetilo desenrollaba
el ADN y activaba el gen, mientras que quitarlo lo volvía a enrollar, inactivando el gen.
O sea, que estamos diciendo que la epigenética es capaz de, partiendo de una misma información
genética de un mismo ADN, cambiar la forma en la que ESTE SE LEE simplemente añadiendo
o quitando ciertas moléculas.
Y ESTO nos lleva de vuelta a la historia de antes, al ejemplo de las gemelas: por mucho
que dos personas nazcan con la misma información genética, nunca van a ser iguales, porque
su epigenética va a ser distinta.
Nunca estarán expresando los mismos genes en cada momento de su vida.
Esto es lo que han visto en multitud de estudios al comparar la metilación del ADN o de la
acetilación de las histonas en las células de gemelos idénticos y ver que efectivamente
existían diferencias epigenéticas entre ellos ya desde su infancia.
No solo eso sino que cuanto más mayores son los gemelos, menos se parecen.
Y no hablamos solo de diferencias físicas, evidentemente, sino también en el desarrollo
de ciertas enfermedades: se ha visto que los gemelos idénticos presentan de forma distinta
enfermedades como la diabetes tipo 1, la diabetes tipo 2, enfermedades autoinmunes, enfermedades
mentales como el trastorno bipolar y la depresión, o distintos tipos de cáncer.
Por ejemplo, en este estudio se compararon gemelas en las que una de ellas había sido
diagnosticada de cáncer de mama, y se observó que aquella mujer que desarrollaba el cáncer
presentaba una metilación exagerada en un gen concreto: el DOK7.
Esto nos puede servir por ejemplo para diagnosticar un cáncer a tiempo, es decir, DOK7 se podría
utilizar como “biomarcador” que indicase la presencia de un cáncer de mama en el cuerpo.
Y después de todo esto, la pregunta estrella es: ¿Por qué esas diferencias?
¿Qué condiciona que la epigenética evolucione de forma distinta en dos personas a priori
idénticas?
Pues aquí viene lo interesante.
Precisamente la epigenética podría explicar la influencia que tiene el entorno en nuestras
características o nuestra salud, es decir, el por qué cosas como la dieta, hacer o no
ejercicio, estar expuestos a ciertas sustancias o tener ciertas infecciones, pueden cambiar
la forma en la que se comportan nuestros genes y por tanto influir en la probabilidad de
padecer ciertas enfermedades.
Pero es que la cosa va más allá: como hemos dicho la epigenética es reversible,
con lo que si conocemos qué cambios epigenéticos se encuentran en determinadas enfermedades
como el cáncer, podríamos llegar a desarrollar fármacos que actúen específicamente sobre
esos cambios para revertir la enfermedad, cosa que ya se está haciendo hoy en día.
O sea, que hemos pasado de soñar con algo tan GRANDE como la EDICIÓN GENÉTICA especialmente
gracias a técnicas tan recientes como el CRISPR, a soñar con algo incluso más específico:
la edición EPIGENÉTICA.
La epigenética es un tema fascinante porque nos enseña una vez más lo maravillosamente
complejo que es nuestro organismo, y cómo por mucho que logremos hasta secuenciar toda
la información genética que se esconde en nuestro ADN, hay algo incluso más allá que
hasta hace unos pocos años ni siquiera hubiéramos imaginado.
Me recuerda a la frase de Manel Esteller, Director del Instituto de Investigación Josep Carreras e investigador de la epigenética del cáncer en la que dice: «Se decía que el ADN era el libro de la vida, pero le faltaban las comas, los puntos, es decir, la epigenética».
Me he dejado algunos puntos como la herencia de la epigenética,




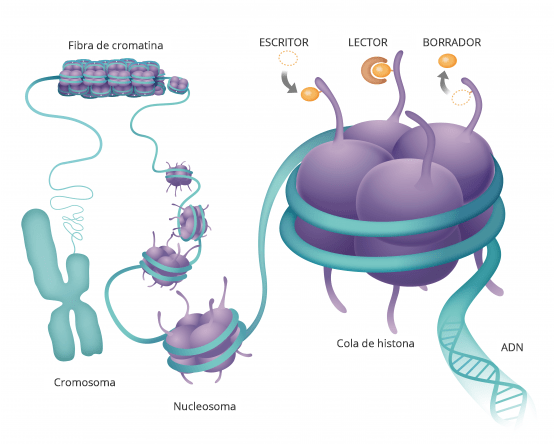
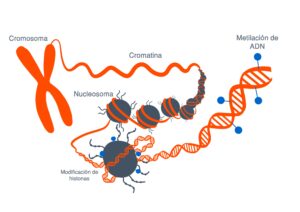 Los mecanismos epigenéticos, que regulan la expresión de los genes sin modificar la secuencia de ADN también pueden contribuir al desarrollo de enfermedades. Imagen: Rosario García, Genotipia.
Los mecanismos epigenéticos, que regulan la expresión de los genes sin modificar la secuencia de ADN también pueden contribuir al desarrollo de enfermedades. Imagen: Rosario García, Genotipia.
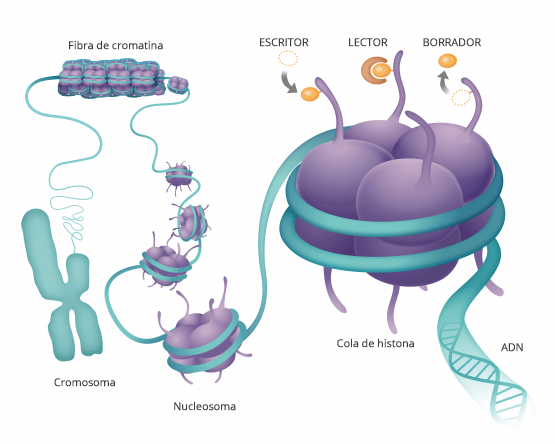
 Abreviar la estructura de un ordenador y evitar así el enorme cableado que desde el procesador central y por la placa base llega a los distintos componentes del ordenador, no solo ahorraría tamaño en este artefacto sino energía rapidez en la comunicación y economía simplemente quitando los cables de Unión y sustituyendolos por Torres receptoras de lo elaborado en el procesador centrar y en los sistemas operativos.
Abreviar la estructura de un ordenador y evitar así el enorme cableado que desde el procesador central y por la placa base llega a los distintos componentes del ordenador, no solo ahorraría tamaño en este artefacto sino energía rapidez en la comunicación y economía simplemente quitando los cables de Unión y sustituyendolos por Torres receptoras de lo elaborado en el procesador centrar y en los sistemas operativos.



![4: Glioblastoma con un alto grado de densidad celular y algunas células multinucleadas [29].](http://enriquerubio.net/wp-content/uploads/4-glioblastoma-con-un-alto-grado-de-densidad-celu.png)
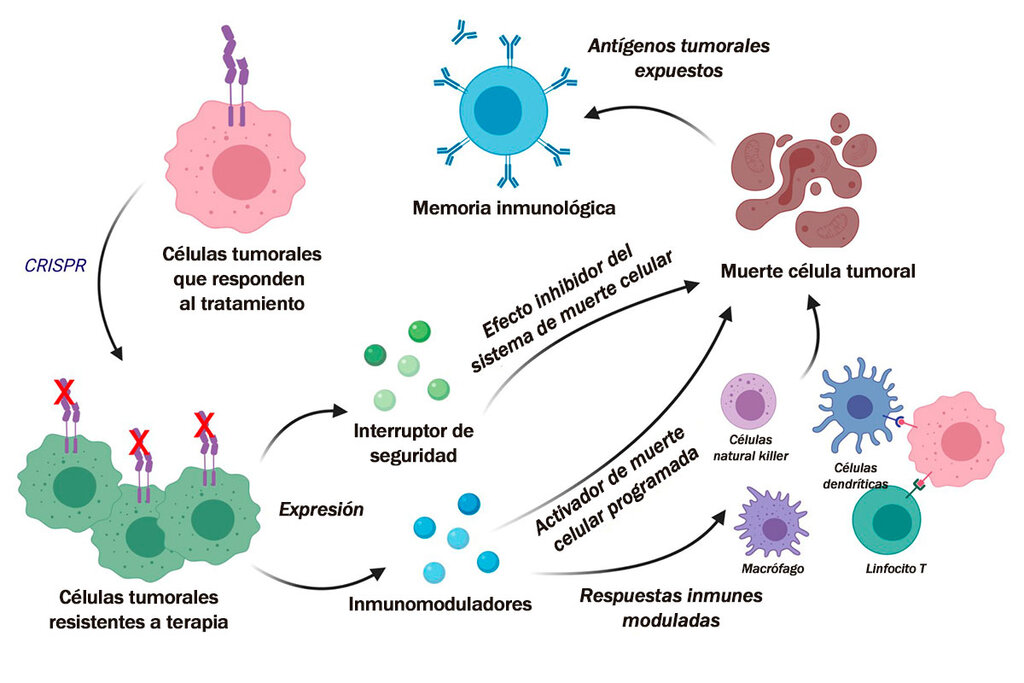 Esquema gráfico del desarrollo de esta nueva terapia celular frente al cáncer. Gráfico: KOK SIONG CHEN Y KHALID SHAH.
Esquema gráfico del desarrollo de esta nueva terapia celular frente al cáncer. Gráfico: KOK SIONG CHEN Y KHALID SHAH.

 Chase Beisel y Oleg Dmytrenko, investigadores de la Universidad de Würzburg, en Alemania. Foto: HIR.
Chase Beisel y Oleg Dmytrenko, investigadores de la Universidad de Würzburg, en Alemania. Foto: HIR.