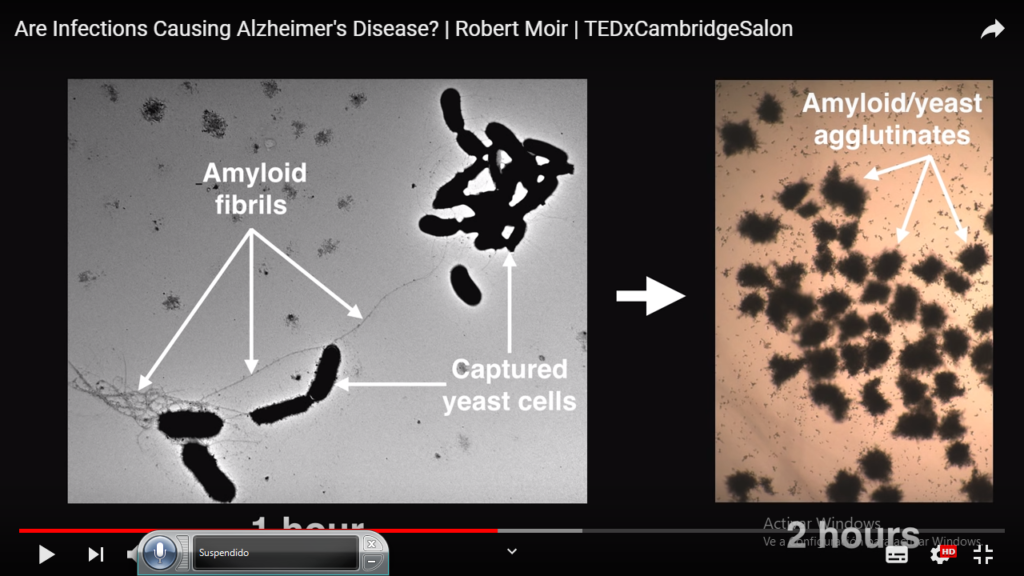
He tenido ocasión de vivir muchos, años con un medico intimo amigo mío que era una especie, de listo y despistado.
Fue siempre un autista social, moderado a buen estudiante y un buen profesional. Era un buen medico y mas de una vez nos salvo a un enfermo. Pero sus despistes eran tan evidentes, que era tratado indistintamente como sabio y tonto. Se caso con una mujer guapísima, con la que tuvo 9 hijos. Y ella giro en torno a él. Su capacidad de introducir innovaciones en medicina, eran manifiesta así como la de hacer buenos negocios con las artes y al mismo tiempo de un desprendimiento y bondad con todo el mundo. Nunca cuido el intelecto y sus amigos preferentes eran gente no recomendable. Como amigo, era magnifico.
Siendo ya muy mayores y en una cena, se rompió todo.
Ya venia fallando en su conducta y sus compañeros, empezaban a alarmarse y atribuirlo estupidamente al alcohol.
Tenia trastornos de la marcha, incontinencia de orina , un discreto despiste.
Una TAC mostro que tenia una enorme hidrocefalia por estenosis congénita del acueducto de Silvio. Una serie de derivaciones del liquido cefalo raquideo, no consiguieron mejorarlo.
Tuvieron que pasar 50 años de conocerlo, hasta que una noche me diera cuenta que su marcha y deterioros sociales no eran normales y si evolutivos. Mi querido amigo se fue y nunca me perdone, no haberme dado cuenta de la organicidad de sus cosas.
Yo no se si esto me motivo a entusiasmarme por los SAVAN, pero ahora los sigo al menos en la literatura, con mucha atención.
El término «idiot savant» («idiota erudito» en francés) fue utilizado por primera vez para describir la condición en 1887 por el médico británico John Langdon Down, conocido por su descripción del síndrome de Down. El término «sabio idiota» se consideró posteriormente erróneo, puesto que no todos los casos reportados se ajustaban a la definición de idiota, originalmente utilizada para una persona con una discapacidad intelectual muy severa. El término «sabio autista» también se utilizó como descripción del trastorno. Pero al igual que «sabio idiota», el término llegó a ser considerado inapropiado porque solo la mitad de los diagnosticados con el síndrome del sabio eran autistas. La necesidad de precisión en el diagnóstico y de no afectar a la dignidad de los afectados,
Sufren desórdenes mentales y discapacidades físicas, mentales o motrices, pero “a cambio” poseen habilidades mentales increíbles. .
Benjamín Rush describió el síndrome de savant por primera vez en 1789. Vio un paciente que era capaz de calcular la edad de las personas tan solo observándolas durante unos segundos.
No siempre estos sabios tienen desordenes de conducta o intelectuales y por ello me preocupo de tres casos que me ha impresionada.
El mas significativo.
Kim Peek: que inspiro la película Rain Man
Nació con macrocefalia, una malformación permanente en el cerebelo, y agenesia en el cuerpo calloso.
Esto le convirtió en una persona muy dependiente, incapaz de realizar tareas básicas, como abrocharse una camisa. Sin embargo, sorprendió al mundo entero con sus portentosas capacidades intelectuales. Tenía una de las memorias más extraordinarias que la ciencia ha podido datar.
Fue capaz de aprenderse cerca de los 8.000 libros que había leído y podía leer dos páginas al mismo tiempo, una con cada ojo. Además, reproducía cosas habiéndolas escuchado o leído tan solo una vez. Llegó a saberse de memoria todos los mapas de Estados Unidos, de manera que aunque no hubiera hecho nunca un determinado recorrido, podía realizarlo sin necesidad de indicaciones o señales.
Su nivel de procesamiento mental era impresionante. Pero, por otro lado, sus limitaciones motrices y cognitivas también eran manifiestas. Por ejemplo, era incapaz de interpretar un poema o inferir conclusiones de una obra. No tenía aptitudes musicales, sin embargo, si escuchaba una canción, podía reproducirla tocando en un piano sin mayor dificultad.
Otro caso es también de una dificultad prodigiosa. Tras un traumatismo se convierte en un superdotado
Jason Padgett: síndrome de savant adquirido
No nació con sus habilidades, sino que estas llegaron cuando tenía 30 años.
Jason era un joven superficial, pero con una conducta normal. Una noche, saliendo con ellos, fue agredido violentamente. Sufrió una conmoción cerebral y, tras pasar por el hospital y volver a casa, se dio cuenta de que todo había cambiado.
Por un lado, empezó a sufrir distintos trastornos como TOC, agorafobia o depresión. Y, por otro lado, llego a ser genial en matemáticas,. Realizaba cálculos mentales y visualizaba la realidad mediante patrones geométricos.
estudiaron al chico y vieron que, tras sufrir la conmoción cerebral, algunas áreas del cerebro que en su día a día permanecían inactivas, con el golpe se activaron para sustituir las funciones dañadas.
Esto es una mentira romántica. Pero si no es verdad, está bien contado.
No tenia estudios, previos, como compararon con registros postraumáticos. Esto es como tantas veces ocurre una mentira para terminar bien.
Pero el caso mas esplendido y delicioso, lo escribe Don Jose Luis Borges, del que dijo “es una larga metáfora del insomnio”.
Después de un día bochornoso, una enorme tormenta color pizarra había escondido el cielo. La alentaba el viento del Sur, ya se enloquecían los árboles; yo tenía el temor (la esperanza) de que nos sorprendiera en un descampado el agua elemental. Corrimos una especie de carrera con la tormenta. Entramos en un callejón que se ahondaba entre dos veredas altísimas de ladrillo. Había oscurecido de golpe; oí rápidos y casi secretos pasos en lo alto; alcé los ojos y vi un muchacho que corría por la estrecha y rota vereda como por una estrecha y rota pared. Recuerdo la bombacha, las alpargatas, era “Funes el memorioso”, Bernardo le gritó imprevisiblemente: ¿Qué hora son Ireneo? Sin consultar el cielo, sin detenerse, el otro respondió: Faltan cuatro minutos para las ocho, joven Bernardo Juan Francisco. La voz era aguda, burlona. Yo soy tan distraído que el diálogo que acabo de referir no me hubiera llamado la atención si no lo hubiera recalcado mi primo, a quien estimulaban (creo) cierto orgullo local, y el deseo de mostrarse indiferente a la réplica tripartita del otro. Me dijo que el muchacho del callejón era un tal Ireneo Funes, mentado por algunas rarezas como la de no darse con nadie y la de saber siempre la hora, como un reloj. Agregó que era hijo de una planchadora del pueblo, María Clementina Funes, y que algunos decían que su padre era un médico del saladero, un inglés O’Connor, y otros un domador o rastreador del departamento del Santo. Vivía con su madre, a la vuelta de la quinta de los Laureles. Los ochenta y cinco y ochenta y seis veraneamos en la ciudad de Montevideo. El ochenta y siete volví a Fray Bentos. Pregunté, como es natural, por todos los conocidos y, finalmente, por el “cronométrico Funes”. Me contestaron que lo había volteado un redomón en la estancia de San Francisco, y que había quedado tullido, sin esperanza. Me dijeron que no se movía del catre, puestos los ojos en la higuera del fondo o en una telaraña. En los atardeceres, permitía que lo sacaran a la ventana. Llevaba la soberbia hasta el punto de simular que era benéfico el golpe que lo había fulminado…
Ireneo, en su rancho de las orillas, no tardó en enterarse del arribo de esos libros anómalos. Me dirigió una carta florida y ceremoniosa, en la que recordaba nuestro encuentro, desdichadamente fugaz, “del siete de febrero del ochenta y cuatro”, ponderaba los gloriosos servicios que don Gregorio Haedo, mi tío, finado ese mismo año, “había prestado a las dos patrias en la valerosa jornada de Ituzaingó”, y me solicitaba el préstamo de cualquiera de los volúmenes, acompañado de un diccionario “para la buena inteligencia del texto original, porque todavía ignoro el latín”.
Arribo, ahora, al más difícil punto de mi relato. Éste (bueno es que ya lo sepa el lector) no tiene otro argumento que ese diálogo de hace ya medio siglo. No trataré de reproducir sus palabras, irrecuperables ahora. Prefiero resumir con veracidad las muchas cosas que me dijo Ireneo. El estilo indirecto es remoto y débil; yo sé que sacrifico la eficacia de mi relato; que mis lectores se imaginen los entrecortados períodos que me abrumaron esa noche. Ireneo empezó por enumerar, en latín y español, los casos de memoria prodigiosa registrados por la Naturalis historia; Ciro, rey de los persas, que sabía llamar por su nombre a todos los soldados de sus ejércitos; Mitríades Eupator, que administraba la justicia en los 22 idiomas de su imperio; Simónides, inventor de la mnemotecnia; Metrodoro, que profesaba el arte de repetir con fidelidad lo escuchado una sola vez. Con evidente buena fe se maravilló de que tales casos maravillaran. Me dijo que antes de esa tarde lluviosa en que lo volteó el azulejo, él había sido lo que son todos los cristianos: un ciego, un sordo, un abombado, un desmemoriado. (Traté de recordarle su percepción exacta del tiempo, su memoria de nombres propios; no me hizo caso.) Diez y nueve años había vivido como quien sueña: miraba sin ver, oía sin oír, se olvidaba de todo, de casi todo. Al caer, perdió el conocimiento; cuando lo recobró, el presente era casi intolerable de tan rico y tan nítido, y también las memorias más antiguas y más triviales. El hecho apenas le interesó. Razonó (sintió) que la inmovilidad era un precio mínimo. Ahora su percepción y su memoria eran infalibles.
Nosotros, de un vistazo, percibimos tres copas en una mesa; Funes, todos los vástagos y racimos y frutos que comprende una parra. Sabía las formas de las nubes australes del amanecer del treinta de abril de mil ochocientos ochenta y dos y podía compararlas en el recuerdo con las vetas de un libro en pasta española que sólo había mirado una vez y con las líneas de la espuma que un remo levantó en el Río Negro la víspera de la acción del Quebracho. Esos recuerdos no eran simples; cada imagen visual estaba ligada a sensaciones musculares, térmicas, etc. Podía reconstruir todos los sueños, todos los entresueños. Dos o tres veces había reconstruido un día entero; no había dudado nunca, pero cada reconstrucción había requerido un día entero. Me dijo: Más recuerdos tengo yo solo que los que habrán tenido todos los hombres desde que el mundo es mundo. Y también: Mis sueños son como la vigilia de ustedes. Y también, hacia el alba: Mi memoria, señor, es como vaciadero de basuras. Una circunferencia en un pizarrón, un triángulo rectángulo, un rombo, son formas que podemos intuir plenamente; lo mismo le pasaba a Ireneo con las aborrascadas crines de un potro, con una punta de ganado en una cuchilla, con el fuego cambiante y con la innumerable ceniza, con las muchas caras de un muerto en un largo velorio. No sé cuántas estrellas veía en el cielo.
La voz de Funes, desde la oscuridad, seguía hablando. Me dijo que hacia 1886 había discurrido un sistema original de numeración y que en muy pocos días había rebasado el veinticuatro mil. No lo había escrito, porque lo pensado una sola vez ya no podía borrársele. Su primer estímulo, creo, fue el desagrado de que los treinta y tres orientales requirieran dos signos y tres palabras, en lugar de una sola palabra y un solo signo. Aplicó luego ese disparatado principio a los otros números. En lugar de siete mil trece, decía (por ejemplo) Máximo Pérez; en lugar de siete mil catorce, El Ferrocarril; otros números eran Luis Melián Lafinur, Olimar, azufre, los bastos, la ballena, el gas, la caldera, Napoleón, Agustín de Vedia. En lugar de quinientos, decía nueve. Cada palabra tenía un signo particular, una especie de marca; las últimas eran muy complicadas…Yo traté de explicarle que esa rapsodia de voces inconexas era precisamente lo contrario de un sistema de numeración. Le dije que decir 365 era decir tres centenas, seis decenas, cinco unidades; análisis que no existe en los “números” El Negro Timoteo o manta de carne.
Había aprendido sin esfuerzo el inglés, el francés, el portugués, el latín. Sospecho, sin embargo, que no era muy capaz de pensar. Pensar es olvidar diferencias, es generalizar, abstraer. En el abarrotado mundo de Funes no había detalles, casi inmediatos. La recelosa claridad de la madrugada entró por el patio de tierra. Entonces vi la cara de la voz que toda la noche había hablado. Ireneo tenía diecinueve años; había nacido en 1868; me pareció monumental como el bronce, más antiguo que Egipto, anterior a las profecías y a las pirámides. Pensé que cada una de mis palabras (que cada uno de mis gestos) perduraría en su implacable memoria; me entorpeció el temor de multiplicar ademanes inútiles. Ireneo Funes murió en 1889, de una congestión pulmonar. 1942 .
Qué duda cabe qué Funes tenía el patrón de SAVANT, pero Borges, sin ser científico , lo describe como hacen los poetas con más belleza .
y qué decir de la biología de este síndrome, sería correcto decir “no lo sé”. Todo lo que se ha dicho hasta ahora, no encaja claro, que no hace falta que el cerebro esté integro morfológicamente, para que algunas cualidades sean excepcionales.
Pero es posible los condicionamientos sociales, no se puedan adquirir en cerebros rotos.
El estudio general sobre todo en Ciencias médicas lo hemos hecho desde las partes olvidándonos que todo era un conjunto, y que el estudio de las partes era insuficente.
La genética da para mucho y explica amplia parte de la patología, pero una parte de las enfermedades no tienen su origen en la genética, sino en lo externo y de hecho gran variedad de factores pueden afectar a la salud.
El estudio de los procesos patológicos de forma holística se debe acercar más a lo real.
De aquí que este teniendo progresión lo que se llama
EXPOSOMA.
El término EXPOSOMA engloba todos los factores que no son genéticos y pueden alterar la salud
La publicación en 2001 de los resultados del Proyecto del Genoma Humano marcó un antes y un después en la historia de la Medicina.
Le toca el turno ahora al Exposoma.
El exposoma es un término acuñado en 2005 que engloba “todo aquello que no es genética y puede condicionar el estado de salud y enfermedad”. Son un amplio catálogo de factores, tanto de la esfera social, económica y psicológica a otros como la radiación, la exposición a químicos, la dieta y el ejercicio, el consumo de alcohol, el tabaquismo, el metabolismo hormonal o la microbiota. La contaminación que no es genética en principio, es responsable del 16% de las muertes en el mundo.
Los determinantes no genéticos de las enfermedades, que son en principio mas difíciles de determinar, posiblemente porque se le ha prestado menos atención que a lo genomico, llegara sin duda a ser junto a los genes con los que sin duda se imbricara importante y ayudara en la comprensión de la patología globalmente
La Fundación Instituto Roche celebró este miércoles la III Jornada Anticipando la Medicina del Futuro y entre los temas que se abordaron, un grupo de expertos autores del informe Anticipando sobre Exposoma, analizó el estado actual y los retos del estudio de los condicionantes no genéticos en la enfermedad y la salud. Mientras que la investigación en genómica ha sido liderada por la comunidad biomédica, en el estudio del exposoma intervienen disciplinas tan diversas como la sociología, la toxicología o la salud laboral, entre otras muchas.
Se sabe que los factores no genéticos juegan un papel esencial en el origen de enfermedades como las cardiovasculares, las oncológicas, las respiratorias y las endocrinas. De los factores ambientales, solo la contaminación de agua, aire y suelo se considera responsable del 16% de las muertes a nivel mundial, unas nueve millones al año, según el programa internacional de investigación Global Burden of Disease cuyos resultados publica periódicamente The Lancet.
En los últimos años ha aumentado el interés por la investigación en el exposoma gracias al reconocimiento de la influencia de factores como el calentamiento global y los productos químicos, así como a los avances en la recogida y manejo de los datos. “Ahora podemos acercarnos al estudio de estos factores de una manera global, sistemática, agregada y con una potencia impresionante”.
Como consecuencia de la enorme variabilidad de condicionantes no genéticos, el estudio del exposoma es extremadamente complejo, porque además el exposoma es dinámico, y su efecto sin duda intervendrá en la genética”.
La ventana de susceptibilidad hace referencia a las etapas de la vida en la que se es más vulnerable al exposoma
Olea resalta que un aspecto relevante en el estudio del exposoma es el momento de exposición y la llamada ventana de susceptibilidad. Etapas de especial vulnerabilidad a determinadas exposiciones, que pueden condicionar hitos en el desarrollo y madurez, son la etapa prenatal y la primera infancia, o la adolescencia.
“Aunque el exposoma se define como el conjunto de todas las exposiciones ambientales a lo largo de la vida, es diferente a los 80 años que en la etapa fetal, cuando los órganos se están desarrollando”.
Martine Vrijheid, responsable del programa Infancia y Medio Ambiente de ISGlobal, destaca que el estudio del exposoma es más complejo que el del genoma y precisa de un amplio abanico de herramientas para las mediciones, desde la biomonitorización humana y ambiental (mediante la recogida de muestras), a instrumentos más sencillos como los cuestionarios.
“Lo que queremos hacer en un estudio de exposoma no es solo relacionar una exposición con un efecto o una enfermedad, sino mirar la complejidad de todos estos factores y cómo se relacionan en conjunto”, afirma Vrijheid. En este terreno resalta los avances en estadística y la inteligencia artificial que permiten esta investigación. “Es importante mirar las interacciones entre exposiciones: cómo interactúan si se está expuesto a bisfenol A, se tiene un nivel bajo actividad física y se se está expuesto a un nivel alto de contaminación del aire”.
En los últimos años en investigación del exposoma se ha producido un cambio de paradigma, entiende Argelia Castaño, directora del Centro Nacional de Sanidad Ambiental del Instituto de Salud Carlos III y asesora de la Organización Mundial de la Salud. Mientras que tradicionalmente, cuestiones como la exposición a químicos, “siempre se ha abordado de manera individualizada”, en los últimos años se tiende a una investigación “más holística. No vivimos en una burbuja, con un solo agente causal, sino que estamos inmersos en una mezcla. Todas las herramientas que se están desarrollando y han venido desarrollándose en los últimos 50 años inciden en ello”, alerta Castaño.
En esta línea, Olea asegura que este cambio de mentalidad debe alcanzar a la toxicología regulatoria. Clasificar sustancias químicas de forma individual por su capacidad carcinogénica, mutágénica o tóxica para la reproducción “es de una simpleza que no es admisible”. Al mismo tiempo, señala que “con la múltiple información de pequeños compuestos químicos, en muy bajas dosis, actuando en periodos críticos y con efectos a largo plazo, todo el sistema regulador se ha visto muy cuestionado”.
Un llamamiento de los expertos es que el reconocimiento del exposoma se traslade a la atención sanitaria, en especial a la atención primaria. Sin embargo, Jaime Mendiola, investigador del Ciber de Epidemiología y Salud Pública (Ciberesp), señala que esta traslación comienza a producirse pero solo en las especialidades. Y pone de ejemplo el caso de la Sociedad Americana de Medicina Reproductiva y el Colegio de Obstetras y Ginecólogos “que recomiendan desde hace años que en la anamnesis a sus pacientes por temas de reproducción se les pregunte por temas medioambientales”.
Dada la necesidad que tiene nuestro cerebro de fragmentar el conocimiento porque no lo entiende en conjunto, este nuevo concepto exposoma, me huele, que nos lleva a la medicina total a la medicina general, por estudiar los procesos en su totalidad y entonces terminamos llamándole. “TODOMA”.
Biografía
Naiara Brocal Dom, 20/12/2020
Nicolás Olea, catedrático del Departamento de Radiología y Medicina Física en la Universidad de Granada y coordinador del informe
La edición genética con CRISPR puede mejorar la capacidad de las células CAR-T para eliminar el cáncer de la sangre y está detrás de un tratamiento para hematopatías genéticas.
La técnica CRISPR descubierta por el Profesor Mujica, ya está dando resultados, aunque el premio Nobel de Química 2020, no se lo dieran a el.
Cada día me impresiona más la facilidad que me ofrece los motores de búsqueda en Internet . puedo saber antes que publiquen las revistas prestigiosas , lo que se está haciendo en el mundo de la ciencia .
Con internet concretamente con Google estoy informado de lo que se está haciendo en el mundo científico . Seguro que tiene mucho de fake News , pero no más que las revistas que portan el trabajo original.
Cuál va a ser el futuro de esta forma de informarse, no lo sé , pero sí que es de gran utilidad.
Hace 60 años que soy neurocirujano y en consecuencia tengo miles de revista a las que consultó, con dificultad. Cuesta mucho esfuerzo encontrar un artículo y más aún encontrar aquellos que le están relacionados y que vienen en otras revistas . Todo eso se consigue en internet con mucho menos esfuerzo y mucho menos tiempo. seguro que esto no lo comparten muchos estudiosos, pero los que solo queremos estar informados y saber por donde van los tiros, estamos encantados.
Los que son unos fenómenos, son los divulgadores, no solo investigadores, sino medios de comunicación y en consecuencias periodistas, que lo divulgan y lo pueden leer todo los que le gusta saber, aunque no sean en libros.
Claro que todo esta cambiando y tenemos que adaptarnos, pero esta es una caualidad del hominido. Dejar lo pasado aunque si siempre tenerlo en cuenta y que sirva como puntos de partida.
Estos artículos que ahora copio, tienen ese fundamento.
A mi me impresionan su parecido a una serie de fichas de dominó, que se colocan paralelas y verticalmente, basta mover una de ellas, para que caigan todas.
En biología pasa exactamente lo mismo, una particula que produce un daño, no hace falta eliminarla, con borrar los metabolitos que dan lugar a que actúen, se consigue detener el proceso.
La terapia de células CAR-T desarrollada en la Universidad de Pennsylvania (tisagenlecleucel) induce remisiones importantes en la leucemia linfoblástica aguda (LLA) de células B en niños y en adultos con linfoma B difuso de célula grande, pero no todos los enfermos responden igual de bien. El equipo que diseñó este tratamiento ha ideado una manera, utilizando CRISPR-Cas9, de obtener respuestas en más pacientes y evitar las recaídas.
Con el sistema de edición de genes CRISPR-Cas9 –reconocido con el último Premio Nobel de Química– eliminan el gen CD5 de las células CAR-T. El gen CD5 produce una proteína que puede inhibir la activación de las células encargadas de acabar con el cáncer. Las células CAR-T reforzadas por la edición de CRISPR conseguían mejores resultados en modelos murinos de leucemia, tanto en la remisión del cáncer como en la supervivencia, según expusieron estos investigadores en la reunión anual telemática de la Sociedad Americana de Hematología (ASH).
«Hemos demostrado, por primera vez, que podemos utilizar con éxito CRISPR-Cas9 para eliminar el gen CD5 en las células CAR-T y mejorar su capacidad para atacar el cáncer», dijo el investigador que presentó los resultados, Marco Ruella, de la División de Hematología-Oncología de la Facultad de Medicina Perelman de la Universidad de Pennsylvania y director científico del Programa de Linfoma. «La diferencia entre las células CAR T editadas y no editadas fue sorprendente en varios modelos de cáncer».
El equipo experimentó en ratones con leucemia de células T y B, y linfoma. Los ratones que recibieron células T con el CAR a las que se suprimió CD5 mostraron niveles más altos de proliferación de células T en la sangre periférica, así como una reducción significativa en el tamaño del tumor y mejores resultados de supervivencia en comparación con los ratones a los que se infundieron células CAR-T no editadas.
«Básicamente, en la mayoría de los tipos de cáncer, cuanto menos CD5 se exprese en las células T, mejor será el resultado», ha continuado Ruella. «El nivel de CD5 en las células T es importante».
De hecho, las células CAR-T con deleción de CD5 fueron significativamente más potentes que las células CAR-T sin la deleción (CD5 +) también cuando se dirigían a células B CD19+. Al probarse en modelo de leucemia de células T, esta terapia celular curó a más del 50% de los animales, mientras en modelos de leucemia de células B CD19 + observaron remisiones completas a largo plazo en la mayoría de los animales.
La edición con CRISPR-Cas9 de las células CAR podría «a largo plazo, representar una estrategia más universal para mejorar los efectos antitumorales de las células CAR-T», ha dicho Carl June, director del Centro de Inmunoterapia Celular en el Centro del Cáncer Abramson y uno de los autores del estudio. Este equipo espera iniciar un ensayo clínico de fase I con este enfoque en 2021.
La reunión de la ASH ha sido testigo de la versatilidad de las ‘tijeras genéticas’ CRISPR-Cas9. Además de su aplicación en la inmunoterapia con linfocitos T CAR, se han presentado resultados de un ensayo en el que se usa la edición genética para tratar la anemia de células falciformes y la beta-talasemia.
Con este sistema se edita ex vivo la región potenciadora eritroide de BCL11A en células madre y progenitoras hematopoyéticas. Como resultado, aumenta la hemoglobina fetal, de forma que suple el déficit de esa proteína en pacientes con estas enfermedades hematológicas. Los resultados de este estudio también se publicaron en The New England Journal of Medicine.
Haydar Frangoul, del Hospital Infantil TrisStar Centennial en Nashville (Tennesee), presentó en la reunión además de los datos de seguimiento de dos pacientes -uno con beta-talasemia y otro con anemia de células falciformes- que recoge la revista médica, resultados de otros siete enfermos con beta-talasemia y tres con anemia falciforme.
Antes del tratamiento, los pacientes con beta-talasemia necesitaban transfusiones cada mes y los pacientes con anemia drepanocítica sufrían crisis vasoclusivas con dolor intenso también con regularidad. Después del tratamiento, ninguno de los pacientes con talasemia ha necesitado transfusiones, ni los pacientes con anemia drepanocítica ha experimentado crisis.
Las terapias avanzadas son uno de los campos más dinámicos de investigación en biomedicina.
Las terapias avanzadas ya están cambiando las perspectivas de tratamiento en oncohematología y enfermedades raras, pero lo que se vislumbra es todavía la punta del iceberg de lo que está por llegar.
Bueren ha insistido en que la terapia génica ya está consiguiendo la curación de enfermedades mortales, pero su desarrollo acaba de arrancar y estos éxitos son “pruebas de concepto” de su papel en la medicina del futuro.
Junto con los tumores hematológicos, las terapias avanzadas también están revolucionado el área de enfermedades raras al tiempo que están poniendo contra las cuerdas la sostenibilidad de los sistemas sanitarios. Según Ángel Carracedo, director de la Fundación Pública Gallega de Medicina Genómica (Servicio Gallego de Salud), se calcula que el 10% del gasto farmacéutico global en 2027 corresponderá a tratamientos para enfermedades minoritarias.
De momento las agencias europea (EMA) y americana (FDA) han dado luz verde a seis terapias génicas para enfermedades monogénicas, y hay otras tres aprobadas en oncología. Entre estas, se encuentra la terapia para la atrofia muscular espinal (AME) que con su precio de salida al mercado estadounidense de 2,12 millones de dólares recibió el sobrenombre el medicamento más caro del mundo.
Uno de los retos es conseguir que este tipo de tratamientos alcance otras enfermedades, no tan graves o para las que existe alternativa terapéutica. La terapia génica puede lograr el control de una enfermedad con una única inyección en la vida frente a tratamientos crónicos, ha explicado Gloria González-Aseguinolaza, subdirectora del Centro de Investigación Médica Aplicada (CIMA) de la Universidad de Navarra y directora científica de Vivet Therapeutics. “En enfermedades como la AME está clarísimo, pero en otras como la fenilcetonuria o la enfermedad de Wilson la terapia génica va a llegar, pero tenemos que resolver cuestiones como el coste”.
Dentro de la terapia génica, Lluis Montoliu, investigador científico del Centro Nacional de Biotecnología (CNB), ha resaltado otra revolución, la de la edición génica, aplicable en CAR-T. “Hace unos días se han publicado los primeros resultados espectaculares en anemia falciforme y betatalasemia de pacientes que ya están libres de las transfusiones”.
La terapia celular podría ser útil en distrés respiratorio en covid-19
También en este terreno Montoliu ha observado que queda mucho por aprender y que más allá de la edición génica destinada a inactivar una función de un gen, hay que avanzar en el conocimiento de aquellas aplicaciones de edición que pretenden sustituir una secuencia incorrecta y reemplazarla por lo correcta. “Todavía no sabemos que ese reemplazo ocurra la mayor parte de las veces y no interfiera con otras partes del genoma o del organismo”.
Por otra parte, en el segmento de terapia celular, Bueren ha recordado que el paradigma es el trasplante de progenitores hematopoyéticos pese a que no se considera terapia avanzada al no existir manipulación de las células. Este tipo de aproximación se investiga en un buen número de enfermedades inflamatorias y autoinmunes y en regeneración de tejidos, y se emplea en el tratamiento de fístulas asociadas a Crohn. La terapia celular autorizada en esta indicación tiene firma española y surgió de la investigación de Damián García-Olmo en la Fundación Jiménez Díaz.
Entre los campos en que se considera que la terapia celular puede ser útil están la regeneración ósea y de cartílago, la córnea, el miocardio, la piel y las enfermedades neurodegenerativas. Asimismo en artritis reumatoide, diabetes e incluso el distrés respiratorio en covid-19.
La terapia génica lleva este mismo camino
El primer paciente tratado en ensayo con la terapia génica para la deficiencia en piruvato quinasa alcanza buenos resultados preliminares.
Cultivos celulares utilizados en el ensayo llevado a cabo en la Fundación Jiménez Díaz (Madrid).
La terapia génica puede cambiar el rumbo terapéutico de ciertas enfermedades hematológicas, al actuar sobre el origen de la patología, el defecto genético.
Es el caso de la deficiencia en piruvato quinasa (PKD) es un trastorno hematológico monogénico con una prevalencia estimada de 3.000 a 8.000 pacientes en los Estados Unidos y la Unión Europea.
Resulta de una mutación en el gen PKLR, codificador para la enzima piruvato quinasa, a su vez un componente clave de la vía glucolítica de los glóbulos rojos. Las manifestaciones clínicas de la afección varían de anemia leve a potencialmente mortal. Los niños son el subgrupo más gravemente afectado; entre los tratamiento se incluyen esplenectomía y transfusiones de glóbulos rojo
Ahora, por primera vez, el empleo de terapia génica ha conseguido en un paciente buenos resultados a los tres meses de seguimiento.
El tratamiento es un medicamento huérfano desarrollado por el consorcio del Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas (CIEMAT), el Centro de Investigación Biomédica en Red de Enfermedades Raras (CiberER) y el Instituto de Investigación Sanitaria de la Fundación Jiménez Díaz (IIS-FJD), que se ha demostrado bien tolerado y seguro. En el ensayo, ha incrementado los valores de hemoglobina y otros parámetros característicos de la hemólisis característica de estos pacientes a valores de normalidad, con lo que el paciente tratado ha quedado independiente de transfusiones.
El medicamento desarrollado, designado como medicamento huérfano por las agencias reguladoras europea y americana (EMA y FDA), ha sido el resultado del trabajo de más de 15 años coordinado por José Carlos Segovia, jefe de la Unidad de Diferenciación y Citometría, de la División de Terapias innovadoras dirigida por Juan Bueren, del Departamento de Investigación Básica del CIEMAT.
El tratamiento consiste en una modificación ex vivo de los progenitores hematopoyéticos, mediada por vectores lentivirales, que ha sido llevado a cabo en las instalaciones de la Fundación Jiménez Díaz por José Luis López Lorenzo, coordinador de la Unidad de Trasplante Hematopoyético del Servicio de Hematología de este hospital, e investigador principal del ensayo clínico en pacientes adultos.
Segovia se ha mostrado esperanzado, pero cauto con los resultados obtenidos en el ensayo: “Todavía es pronto, porque son datos del seguimiento a tres meses, pero si la recuperación que hemos visto, se mantiene a largo plazo, sí podríamos hablar de una cura de la enfermedad con un único tratamiento. Hemos observado una recuperación a niveles normales de los valores de hemoglobina. Ahora nos queda esperar y confiar en que se sigan manteniendo esos niveles”.
El desarrollo de este tratamiento es fruto de tres tesis doctorales, presentadas por los ya doctores Néstor W. Meza (2005), María García Gómez (2012) y Sergio López Manzaneda (2016). El desarrollo a nivel clínico ha sido posible gracias a una estrecha colaboración público-privada con la empresa biofarmacéutica Rocket Pharmaceuticals, con sede en Nueva York (Estados Unidos), que ha puesto los recursos humanos, técnicos y económicos necesarios para poder llevar el medicamento desarrollado al ensayo clínico. También ha contado con el apoyo importante de la Fundación Botín.
Los datos del ensayo se han presentado en la reunión anual de la Sociedad Americana de Hematología (ASH), celebrada en formato telemático, y en la que se han presentado otros estudios esperanzadores con terapia génica. En concreto, y coincidiendo con la publicación en The New England Journal of Medicine, se aportaron los resultados de seguimiento de varios pacientes con beta-talasemia y anemia de células falciformes. El tratamiento administrado también consistió en la edición ex vivo de la región potenciadora eritroide de BCL11A en células madre y progenitores hematopoyéticos. Como resultado, aumentó en los pacientes la hemoglobina fetal, supliendo así el déficit de esta proteína.
La edición genética en esos casos se llevó a cabo mediante las herramientas CRISPR-Cas9, algo que los científicos españoles también están explorando. Como expone Segovia, en este congreso “hemos presentado una comunicación oral sobre la corrección genética de la deficiencia en piruvato quinasa mediante lo que podría llamarse terapia génica 2.0”. La técnica de CRISPR-Cas9 permite hacer la corrección en el locus mutado y favorecer así la integración de la copia correcta de manera específica y regulada por las secuencias endógenas“.
Este grupo del CIEMAT combina la edición génica de CRISPR-Cas9 –mediante la que realizan la ‘rotura’ en el lugar deseado- con vectores de virus adeno-asociados (AAV), que emplean para introducir las secuencias correctas en las células.
Mientras se asienta esta nueva estrategia, el científico ha comentado que el ensayo que están llevando a cabo con la PKD abre la puerta a aplicar la terapia génica en otras anemias hemolíticas raras. “Estamos trabajando ya con la anemia diseritropoyética congénita tipo II. Esta enfermedad resulta de mutaciones en el gen SEC23B, que está implicado en el tráfico de proteínas de la célula y de manera específica en el linaje de los eritrocitos”. La afección causa anemia grave que puede llegar a requerir un trasplante alogénico de médula ósea.
Cajal mejoró el método de Golgi y comenzó a estudiar embriones de pollos y otros animales del jardín. formuló la teoría de la neurona que se basa en tres pilares:
Las neuronas son células individuales y no un continuo.
Las neuronas se comunican entre si en sitios concretos (llamados sinapsis por Sherrington).
Principio de la polarización dinámica. El flujo de corriente va desde las dendritas (entrada) hasta el axón (salida).
Hasta aquí la revolución de CaJal, el sistema nerviosos no es un retidulo continuo, esta compuesto por células. Fundandose en esto. Charles Scott Sherrington y colaboradores, describen la Sinapsis, que vienen de sinapteína, que se forman con las palabras griegas sin-, que significa «juntos», y hapteina, «con firmeza».
La sinapsis (del griego ύναψις [sýnapsis] [«neurotrasmisores»], ‘unión’, ‘enlace’1) es una aproximación (funcional) intercelular especializada entre neuronas,2 ya sean entre dos neuronas de asociación, una neurona y una célula receptora o entre una neurona y una célula efectora (casi siempre glandular o muscular). En estos contactos se lleva a cabo la transmisión del impulso nervioso.
Desde el punto de vista histológico y funcional, una neurona tiene tres zonas principales: el cuerpo o soma, las dendritas y el axón
Las conexiones pueden establecerse a muy corto alcance, a unos cientos de micrómetros a la redonda, o a distancias mucho mayores.).
Una sinapsis prototípica, como las que aparecen en los botones dendríticos, consiste en unas proyecciones citoplasmáticas con forma de hongo desde cada célula que, al juntarse, los extremos de ambas se aplastan uno contra otro. En esta zona, las membranas celulares de ambas células se juntan en una unión estrecha que permite a las moléculas de señal llamadas neurotransmisores pasar rápidamente de una a otra célula por difusión. El canal de unión de la neurona postsináptica es de aproximadamente 20 nm de ancho, y se conoce como hendidura sináptica.
Estas sinapsis son asimétricas tanto en su estructura como en su funcionamiento. Sólo la neurona presináptica segrega los neurotransmisores, que se unen a los receptores transmembrana que la célula postsináptica tiene en la hendidura. El terminal nervioso presináptico (también llamado botón sináptico o botón) normalmente emerge del extremo de un axón, mientras que la zona postsináptica normalmente corresponde a una dendrita, al cuerpo celular o a otras zonas celulares. La zona de la sinapsis donde se libera el neurotransmisor se denomina zona activa. En las zonas activas, las membranas de las dos células adyacentes están unidas estrechamente mediante proteínas de adhesión celular. Justo tras la membrana de la célula postsináptica aparece un complejo de proteínas entrelazadas denominado densidad postsináptica. Las proteínas de la densidad postsináptica cumplen numerosas funciones, que van desde el anclaje y movimiento de receptores de neurotransmisores de la membrana plasmática, hasta el anclaje de varias proteínas reguladoras de la actividad de estos receptores.
Tipos de sinapsis]
Sinapsis eléctrica
Es aquella en la que la transmisión entre la primera neurona y la segunda no se produce por la secreción de un neurotransmisor, como en las sinapsis químicas (véase más abajo), sino por el paso de iones de una célula a otra a través de uniones gap, pequeños canales formados por el acoplamiento de complejos proteicos, basados en conexiones, en células estrechamente adheridas.
La sinapsis eléctrica es la más común en los vertebrados menos complejos y en algunos lugares del cerebro de los mamíferos. Son más rápidas que las sinapsis químicas pero menos plásticas; por lo demás, son menos propensas a alteraciones o modulación porque facilitan el intercambio entre los citoplasmas de iones y otras sustancias químicas. En los vertebrados son comunes en el corazón y el hígado.
Las sinapsis eléctricas tienen tres ventajas muy importantes:
La sinapsis eléctrica posee una transmisión bidireccional de los potenciales de acción, en cambio la sinapsis química solo posee la comunicación correccional.
En la sinapsis eléctrica hay una sincronización en la actividad neuronal, lo cual hace posible una acción coordinada entre ellas.
La comunicación es más rápida en la sinapsis eléctrica que en la química, debido a que los potenciales de acción pasan a través del canal proteico directamente sin necesidad de la liberación de los neurotransmisores.
Sinapsis química
La sinapsis química se establece entre células que están separadas entre sí por un espacio de unos 20-30 nanómetros (nm), la llamada hendidura sináptica.
La liberación de neurotransmisores es iniciada por la llegada de un impulso nervioso (o potencial de acción), y se produce mediante un proceso muy rápido de secreción celular: en el terminal nervioso presináptico, las vesículas que contienen los neurotransmisores permanecen ancladas y preparadas junto a la membrana sináptica. Cuando llega un potencial de acción se produce una entrada de iones calcio a través de los canales de calcio dependientes de voltaje. Los iones de calcio inician una cascada de reacciones que terminan haciendo que las membranas vesiculares se fusionen con la membrana presináptica y liberando su contenido a la hendidura sináptica. Los receptores del lado opuesto de la hendidura se unen a los neurotransmisores y fuerzan la apertura de los canales iónicos cercanos de la membrana postsináptica, haciendo que los iones fluyan hacia o desde el interior, cambiando el potencial de membrana local. El resultado es excitatorio en caso de flujos de despolarización, o inhibitorio en caso de flujos de hiperpolarización. El que una sinapsis sea excitatoria o inhibitoria depende del tipo o tipos de iones que se canalizan en los flujos postsinápticos, que a su vez es función del tipo de receptores y neurotransmisores que intervienen en la sinapsis.
La suma de los impulsos excitatorios e inhibitorios que llegan por todas las sinapsis que se relacionan con cada neurona (1000 a 200 000) determina si se produce o no la descarga del potencial de acción por el axón de esa neurona.
Se distinguen tres tipos principales de transmisión sináptica; los dos primeros mecanismos constituyen las fuerzas principales que rigen en los circuitos neuronales:
transmisión excitadora: aquella que incrementa la posibilidad de producir un potencial de acción;
transmisión inhibidora: aquella que reduce la posibilidad de producir un potencial de acción;
transmisión moduladora: aquella que cambia el patrón y/o la frecuencia de la actividad producida por las células involucradas.
.
Generalmente, si una sinapsis excitatoria es fuerte, un potencial de acción en la neurona presináptica iniciará otro potencial en la célula postsináptica. En una sinapsis débil, el potencial excitatorio postsináptico («PEPS») no alcanzará el umbral para la iniciación del potencial de acción. En el cerebro, cada neurona mantiene conexiones o sinapsis con muchas otras, pudiendo recibir cada una de ellas múltiples señales. Cuando se disparan potenciales de acción simultáneamente en varias neuronas que se unen en sinapsis débiles a otra neurona, pueden forzar el inicio de un impulso en esa célula a pesar de que las sinapsis son débiles.
Una neurona presináptica que libera neurotransmisores inhibitorios, como el GABA, puede generar un potencial inhibitorio postsináptico («PIPS») en la neurona postsináptica, bajando su sensibilidad y la probabilidad de que se genere un potencial de acción en ella. Así la respuesta de una neurona depende de las señales que recibe de otras, con las que puede tener distintos grados de influencia, dependiendo de la fuerza de la sinapsis con esa neurona. John Carew Eccles realizó algunos experimentos importantes en los inicios de la investigación sináptica, por los que recibió el Premio Nobel de Fisiología o Medicina en 1963.
Tras la fusión de las vesículas sinápticas y la liberación de las moléculas transmisoras en la hendidura sináptica, el neurotransmisor es rápidamente eliminado del espacio por proteínas especializadas en su reciclaje, situadas en las membranas tanto presináptica como postsináptica. Esta recaptación evita la desensibilización de los receptores postsinápticos y asegura que los potenciales de acción subsiguientes generen un PEP de la misma intensidad. La necesidad de una recaptación y el fenómeno de la desensibilización en los receptores y canales iónicos significa que la fuerza de la sinapsis puede disminuir si un tren de potenciales de acción llega en una sucesión rápida, un fenómeno que hace que exista una dependencia de la frecuencia en las sinapsis. El sistema nervioso se aprovecha de esta propiedad para computaciones, y puede ajustar las sinapsis mediante la fosforilación de las proteínas implicadas. El tamaño, número y tasa de reposición de las vesículas también está sujeto a regulación, así como otros muchos aspectos de la transmisión sináptica. Por ejemplo, un tipo de fármaco conocido como inhibidores selectivos de la recaptación de serotonina o SSRI afectan a ciertas sinapsis inhibiendo la recaptación del neurotransmisor serotonina. Por el contrario, un neurotransmisor excitatorio muy importante, la acetilcolina, no es recaptada, pero es eliminada por acción de la enzima acetilcolinesterasa.
La modificación de los parámetros sinápticos pueden modificar el comportamiento de los circuitos neurales y la interacción entre los diferentes módulos que componen el sistema nervioso (modal). Dichos cambios están englobados en un fenómeno conocido como neuroplasticidad o plasticidad neuronal.
El lenguaje químico del cerebro
Foto: Paweł Czerwiński en Unsplash.
A su vez, en el proceso actúan proteínas que hacen posible la sinapsis, conformando el proteoma sináptico.
Un nuevo estudio realizado por investigadores del Instituto de Ciencia y Tecnología de Okinawa, en Japón, descifra el lenguaje químico ligado al proteoma sináptico y revela su importancia en las redes cerebrales que favorecen la memoria, el aprendizaje, la atención o la ubicación espacial.
Según un artículo publicado en Medical Xpress, comprender este fascinante lenguaje molecular es de vital importancia por muchas razones, pero principalmente porque las fallas en el proceso sináptico forman parte de la raíz de una gran cantidad de enfermedades cerebrales, como el autismo, el Alzheimer, la epilepsia, el Parkinson o la esquizofrenia, entre otras.
La investigación de los especialistas japoneses, publicada en la revista Proceedings of the National Academy of Sciences (PNAS), podría favorecen nuevos abordajes y tratamientos al facilitar la comprensión del rol que juegan las proteínas en el circuito comunicacional del cerebro, como así también en su conexión con el resto del cuerpo.
Según el Dr. Zacharie Taoufiq, autor principal del estudio, “esta investigación ha dado como resultado un catálogo de todas las diferentes proteínas que participan en las sinapsis. Gracias a esta información contaremos con una gran base para estudiar la diversidad regional y evolutiva del cerebro a nivel sináptico. También será clave para encontrar la causa molecular de la enfermedad de cada paciente, una difícil tarea que nos espera en el futuro”, indicó.
Uno de los aspectos centrales de la investigación, en la que también participaron científicos del Instituto Max Planck de Química Biofísica en Göttingen, Alemania, y de la Universidad de Doshisha en Kioto, Japón, es el reconocimiento y caracterización de las llamadas vesículas sinápticas (SV). Se trata de complejos centros de procesamiento molecular y químico, que funcionan en el marco de una delicada interacción armónica para garantizar una correcta neurotransmisión.
Hasta el momento no se disponía de los datos relativos a la base molecular completa de las sinapsis, pero con la nueva investigación se contará ahora con el relevamiento más extenso y rico de las proteínas presentes en dichos procesos. Para llegar a estos resultados, los investigadores trabajaron en base a un método que les permitió descubrir muchas secuencias ocultas: el objetivo era identificar proteínas que pudieran parecerse en gran medida a otras, pero que presentaran funciones diferentes.
Los resultados superaron las expectativas de los científicos, ya que se hallaron 4.439 proteínas sinápticas, de las cuales 1.466 forman parte de vesículas sinápticas (SV), triplicando el catálogo existente en la actualidad. Al mismo tiempo, descubrieron una gran diversidad en las proteínas SV, que forman subpoblaciones con funciones muy concretas y específicas.
Todo indica que las proteínas implicadas en las sinapsis han desarrollado su propia estructura comunicacional. “Parece que los proteomas sinápticos están estructurados como verdaderos lenguajes, con unas pocas palabras (o proteínas) de uso frecuente y muchos términos menos habituales pero más específicos y significativos «, concluyó el Dr. Taoufiq.
La extensión del catálogo disponible de proteínas sinápticas tiene un valor que excede a su importancia científica, porque permitirá contar con una nueva herramienta para comprender el surgimiento de una gran cantidad de enfermedades cerebrales. Este conocimiento podrá desembocar en alternativas terapéuticas más eficaces, cuando por ejemplo en la actualidad los ensayos clínicos para el tratamiento del Alzheimer alcanzan una tasa de fracaso del 99,6%.
Bibliografía
Bear MF, Connors BW, Paradiso M.A: Neurociencia: explorando el cerebro. Barcelona: Masson, 2002. ISBN 84-458-1259-9.
Hormuzdi SG, Filippov MA, Mitropoulou G, Monyer H, Bruzzone R: «Electrical synapses: a dynamic signaling system that shapes the activity of neuronal networks». Biochim Biophys Acta. 2004 mar 23;1662(1-2):113-37. PMID 15033583.
Kandel ER, Schwartz JH, Jessell TM: Principios de neurociencia. Madrid: McGraw-Hill, 2001, 4.ª ed. ISBN 84-486-0311-7.
Cajal mejoró el método de Golgi para el estudio histologico del sistema nerviosos y comenzó a estudiar embriones de pollos y otros animales del jardín. formuló la teoría de la neurona que se basa en tres pilares:
Las neuronas son células individuales y no un continuo.
Las neuronas se comunican entre si en sitios concretos (llamados sinapsis por Sherrington).
Principio de la polarización dinámica. El flujo de corriente va desde las dendritas (entrada) hasta el axón (salida).
Hasta aquí la revolución de CaJal, el sistema nerviosos no es un retidulo continuo, esta compuesto por células. Fundandose en esto. Charles Scott Sherrington y colaboradores, describen la Sinapsis, que vienen de sinapteína, que se forman con las palabras griegas sin-, que significa «juntos», y hapteina, «con firmeza».
La sinapsis (del griego ύναψις [sýnapsis] [«neurotrasmisores»], ‘unión’, ‘enlace’1) es una aproximación (funcional) intercelular especializada entre neuronas,2 ya sean entre dos neuronas de asociación, una neurona y una célula receptora o entre una neurona y una célula efectora (casi siempre glandular o muscular). En estos contactos se lleva a cabo la transmisión del impulso nervioso.
Desde el punto de vista histológico y funcional, una neurona tiene tres zonas principales: el cuerpo o soma, las dendritas y el axón
Las conexiones pueden establecerse a muy corto alcance, a unos cientos de micrómetros a la redonda, o a distancias mucho mayores.).
Una sinapsis prototípica, como las que aparecen en los botones dendríticos, consiste en unas proyecciones citoplasmáticas con forma de hongo desde cada célula que, al juntarse, los extremos de ambas se aplastan uno contra otro. En esta zona, las membranas celulares de ambas células se juntan en una unión estrecha que permite a las moléculas de señal llamadas neurotransmisores pasar rápidamente de una a otra célula por difusión. El canal de unión de la neurona postsináptica es de aproximadamente 20 nm de ancho, y se conoce como hendidura sináptica.
Estas sinapsis son asimétricas tanto en su estructura como en su funcionamiento. Sólo la neurona presináptica segrega los neurotransmisores, que se unen a los receptores transmembrana que la célula postsináptica tiene en la hendidura. El terminal nervioso presináptico (también llamado botón sináptico o botón) normalmente emerge del extremo de un axón, mientras que la zona postsináptica normalmente corresponde a una dendrita, al cuerpo celular o a otras zonas celulares. La zona de la sinapsis donde se libera el neurotransmisor se denomina zona activa. En las zonas activas, las membranas de las dos células adyacentes están unidas estrechamente mediante proteínas de adhesión celular. Justo tras la membrana de la célula postsináptica aparece un complejo de proteínas entrelazadas denominado densidad postsináptica. Las proteínas de la densidad postsináptica cumplen numerosas funciones, que van desde el anclaje y movimiento de receptores de neurotransmisores de la membrana plasmática, hasta el anclaje de varias proteínas reguladoras de la actividad de estos receptores.
Tipos de sinapsis]
Sinapsis eléctrica
Es aquella en la que la transmisión entre la primera neurona y la segunda no se produce por la secreción de un neurotransmisor, como en las sinapsis químicas (véase más abajo), sino por el paso de iones de una célula a otra a través de uniones gap, pequeños canales formados por el acoplamiento de complejos proteicos, basados en conexiones, en células estrechamente adheridas.
La sinapsis eléctrica es la más común en los vertebrados menos complejos y en algunos lugares del cerebro de los mamíferos. Son más rápidas que las sinapsis químicas pero menos plásticas; por lo demás, son menos propensas a alteraciones o modulación porque facilitan el intercambio entre los citoplasmas de iones y otras sustancias químicas. En los vertebrados son comunes en el corazón y el hígado.
Las sinapsis eléctricas tienen tres ventajas muy importantes:
La sinapsis eléctrica posee una transmisión bidireccional de los potenciales de acción, en cambio la sinapsis química solo posee la comunicación correccional.
En la sinapsis eléctrica hay una sincronización en la actividad neuronal, lo cual hace posible una acción coordinada entre ellas.
La comunicación es más rápida en la sinapsis eléctrica que en la química, debido a que los potenciales de acción pasan a través del canal proteico directamente sin necesidad de la liberación de los neurotransmisores.
Sinapsis química
La sinapsis química se establece entre células que están separadas entre sí por un espacio de unos 20-30 nanómetros (nm), la llamada hendidura sináptica.
La liberación de neurotransmisores es iniciada por la llegada de un impulso nervioso (o potencial de acción), y se produce mediante un proceso muy rápido de secreción celular: en el terminal nervioso presináptico, las vesículas que contienen los neurotransmisores permanecen ancladas y preparadas junto a la membrana sináptica. Cuando llega un potencial de acción se produce una entrada de iones calcio a través de los canales de calcio dependientes de voltaje. Los iones de calcio inician una cascada de reacciones que terminan haciendo que las membranas vesiculares se fusionen con la membrana presináptica y liberando su contenido a la hendidura sináptica. Los receptores del lado opuesto de la hendidura se unen a los neurotransmisores y fuerzan la apertura de los canales iónicos cercanos de la membrana postsináptica, haciendo que los iones fluyan hacia o desde el interior, cambiando el potencial de membrana local. El resultado es excitatorio en caso de flujos de despolarización, o inhibitorio en caso de flujos de hiperpolarización. El que una sinapsis sea excitatoria o inhibitoria depende del tipo o tipos de iones que se canalizan en los flujos postsinápticos, que a su vez es función del tipo de receptores y neurotransmisores que intervienen en la sinapsis.
La suma de los impulsos excitatorios e inhibitorios que llegan por todas las sinapsis que se relacionan con cada neurona (1000 a 200 000) determina si se produce o no la descarga del potencial de acción por el axón de esa neurona.
]
Se distinguen tres tipos principales de transmisión sináptica; los dos primeros mecanismos constituyen las fuerzas principales que rigen en los circuitos neuronales:
transmisión excitadora: aquella que incrementa la posibilidad de producir un potencial de acción;
transmisión inhibidora: aquella que reduce la posibilidad de producir un potencial de acción;
transmisión moduladora: aquella que cambia el patrón y/o la frecuencia de la actividad producida por las células involucradas.
.
Generalmente, si una sinapsis excitatoria es fuerte, un potencial de acción en la neurona presináptica iniciará otro potencial en la célula postsináptica. En una sinapsis débil, el potencial excitatorio postsináptico («PEPS») no alcanzará el umbral para la iniciación del potencial de acción. En el cerebro, cada neurona mantiene conexiones o sinapsis con muchas otras, pudiendo recibir cada una de ellas múltiples señales. Cuando se disparan potenciales de acción simultáneamente en varias neuronas que se unen en sinapsis débiles a otra neurona, pueden forzar el inicio de un impulso en esa célula a pesar de que las sinapsis son débiles.
Una neurona presináptica que libera neurotransmisores inhibitorios, como el GABA, puede generar un potencial inhibitorio postsináptico («PIPS») en la neurona postsináptica, bajando su sensibilidad y la probabilidad de que se genere un potencial de acción en ella. Así la respuesta de una neurona depende de las señales que recibe de otras, con las que puede tener distintos grados de influencia, dependiendo de la fuerza de la sinapsis con esa neurona. John Carew Eccles realizó algunos experimentos importantes en los inicios de la investigación sináptica, por los que recibió el Premio Nobel de Fisiología o Medicina en 1963.
Tras la fusión de las vesículas sinápticas y la liberación de las moléculas transmisoras en la hendidura sináptica, el neurotransmisor es rápidamente eliminado del espacio por proteínas especializadas en su reciclaje, situadas en las membranas tanto presináptica como postsináptica. Esta recaptación evita la desensibilización de los receptores postsinápticos y asegura que los potenciales de acción subsiguientes generen un PEP de la misma intensidad. La necesidad de una recaptación y el fenómeno de la desensibilización en los receptores y canales iónicos significa que la fuerza de la sinapsis puede disminuir si un tren de potenciales de acción llega en una sucesión rápida, un fenómeno que hace que exista una dependencia de la frecuencia en las sinapsis. El sistema nervioso se aprovecha de esta propiedad para computaciones, y puede ajustar las sinapsis mediante la fosforilación de las proteínas implicadas. El tamaño, número y tasa de reposición de las vesículas también está sujeto a regulación, así como otros muchos aspectos de la transmisión sináptica. Por ejemplo, un tipo de fármaco conocido como inhibidores selectivos de la recaptación de serotonina o SSRI afectan a ciertas sinapsis inhibiendo la recaptación del neurotransmisor serotonina. Por el contrario, un neurotransmisor excitatorio muy importante, la acetilcolina, no es recaptada, pero es eliminada por acción de la enzima acetilcolinesterasa.
La modificación de los parámetros sinápticos pueden modificar el comportamiento de los circuitos neurales y la interacción entre los diferentes módulos que componen el sistema nervioso (modal). Dichos cambios están englobados en un fenómeno conocido como neuroplasticidad o plasticidad neuronal.
El lenguaje químico del cerebro
Foto: Paweł Czerwiński en Unsplash.
A su vez, en el proceso actúan proteínas que hacen posible la sinapsis, conformando el proteoma sináptico.
Un nuevo estudio realizado por investigadores del Instituto de Ciencia y Tecnología de Okinawa, en Japón, descifra el lenguaje químico ligado al proteoma sináptico y revela su importancia en las redes cerebrales que favorecen la memoria, el aprendizaje, la atención o la ubicación espacial.
Según un artículo publicado en Medical Xpress, comprender este fascinante lenguaje molecular es de vital importancia por muchas razones, pero principalmente porque las fallas en el proceso sináptico forman parte de la raíz de una gran cantidad de enfermedades cerebrales, como el autismo, el Alzheimer, la epilepsia, el Parkinson o la esquizofrenia, entre otras.
La investigación de los especialistas japoneses, publicada en la revista Proceedings of the National Academy of Sciences (PNAS), podría favorecen nuevos abordajes y tratamientos al facilitar la comprensión del rol que juegan las proteínas en el circuito comunicacional del cerebro, como así también en su conexión con el resto del cuerpo.
Según el Dr. Zacharie Taoufiq, autor principal del estudio, “esta investigación ha dado como resultado un catálogo de todas las diferentes proteínas que participan en las sinapsis. Gracias a esta información contaremos con una gran base para estudiar la diversidad regional y evolutiva del cerebro a nivel sináptico. También será clave para encontrar la causa molecular de la enfermedad de cada paciente, una difícil tarea que nos espera en el futuro”, indicó.
Uno de los aspectos centrales de la investigación, en la que también participaron científicos del Instituto Max Planck de Química Biofísica en Göttingen, Alemania, y de la Universidad de Doshisha en Kioto, Japón, es el reconocimiento y caracterización de las llamadas vesículas sinápticas (SV). Se trata de complejos centros de procesamiento molecular y químico, que funcionan en el marco de una delicada interacción armónica para garantizar una correcta neurotransmisión.
Hasta el momento no se disponía de los datos relativos a la base molecular completa de las sinapsis, pero con la nueva investigación se contará ahora con el relevamiento más extenso y rico de las proteínas presentes en dichos procesos. Para llegar a estos resultados, los investigadores trabajaron en base a un método que les permitió descubrir muchas secuencias ocultas: el objetivo era identificar proteínas que pudieran parecerse en gran medida a otras, pero que presentaran funciones diferentes.
Los resultados superaron las expectativas de los científicos, ya que se hallaron 4.439 proteínas sinápticas, de las cuales 1.466 forman parte de vesículas sinápticas (SV), triplicando el catálogo existente en la actualidad. Al mismo tiempo, descubrieron una gran diversidad en las proteínas SV, que forman subpoblaciones con funciones muy concretas y específicas.
Todo indica que las proteínas implicadas en las sinapsis han desarrollado su propia estructura comunicacional. “Parece que los proteomas sinápticos están estructurados como verdaderos lenguajes, con unas pocas palabras (o proteínas) de uso frecuente y muchos términos menos habituales pero más específicos y significativos «, concluyó el Dr. Taoufiq.
La extensión del catálogo disponible de proteínas sinápticas tiene un valor que excede a su importancia científica, porque permitirá contar con una nueva herramienta para comprender el surgimiento de una gran cantidad de enfermedades cerebrales. Este conocimiento podrá desembocar en alternativas terapéuticas más eficaces, cuando por ejemplo en la actualidad los ensayos clínicos para el tratamiento del Alzheimer alcanzan una tasa de fracaso del 99,6%.
Bibliografía
Bear MF, Connors BW, Paradiso M.A: Neurociencia: explorando el cerebro. Barcelona: Masson, 2002. ISBN 84-458-1259-9.
Hormuzdi SG, Filippov MA, Mitropoulou G, Monyer H, Bruzzone R: «Electrical synapses: a dynamic signaling system that shapes the activity of neuronal networks». Biochim Biophys Acta. 2004 mar 23;1662(1-2):113-37. PMID 15033583.
Kandel ER, Schwartz JH, Jessell TM: Principios de neurociencia. Madrid: McGraw-Hill, 2001, 4.ª ed. ISBN 84-486-0311-7.
Cajal mejoró el método de Golgi y comenzó a estudiar embriones de pollos y otros animales del jardín. formuló la teoría de la neurona que se basa en tres pilares:
Las neuronas son células individuales y no un continuo.
Las neuronas se comunican entre si en sitios concretos (llamados sinapsis por Sherrington).
Principio de la polarización dinámica. El flujo de corriente va desde las dendritas (entrada) hasta el axón (salida).
Hasta aquí la revolución de CaJal, el sistema nerviosos no es un retidulo continuo, esta compuesto por células. Fundandose en esto. Charles Scott Sherrington y colaboradores, describen la Sinapsis, que vienen de sinapteína, que se forman con las palabras griegas sin-, que significa «juntos», y hapteina, «con firmeza».
La sinapsis (del griego ύναψις [sýnapsis] [«neurotrasmisores»], ‘unión’, ‘enlace’1) es una aproximación (funcional) intercelular especializada entre neuronas,2 ya sean entre dos neuronas de asociación, una neurona y una célula receptora o entre una neurona y una célula efectora (casi siempre glandular o muscular). En estos contactos se lleva a cabo la transmisión del impulso nervioso.
Desde el punto de vista histológico y funcional, una neurona tiene tres zonas principales: el cuerpo o soma, las dendritas y el axón
Las conexiones pueden establecerse a muy corto alcance, a unos cientos de micrómetros a la redonda, o a distancias mucho mayores.).
Una sinapsis prototípica, como las que aparecen en los botones dendríticos, consiste en unas proyecciones citoplasmáticas con forma de hongo desde cada célula que, al juntarse, los extremos de ambas se aplastan uno contra otro. En esta zona, las membranas celulares de ambas células se juntan en una unión estrecha que permite a las moléculas de señal llamadas neurotransmisores pasar rápidamente de una a otra célula por difusión. El canal de unión de la neurona postsináptica es de aproximadamente 20 nm de ancho, y se conoce como hendidura sináptica.
Estas sinapsis son asimétricas tanto en su estructura como en su funcionamiento. Sólo la neurona presináptica segrega los neurotransmisores, que se unen a los receptores transmembrana que la célula postsináptica tiene en la hendidura. El terminal nervioso presináptico (también llamado botón sináptico o botón) normalmente emerge del extremo de un axón, mientras que la zona postsináptica normalmente corresponde a una dendrita, al cuerpo celular o a otras zonas celulares. La zona de la sinapsis donde se libera el neurotransmisor se denomina zona activa. En las zonas activas, las membranas de las dos células adyacentes están unidas estrechamente mediante proteínas de adhesión celular. Justo tras la membrana de la célula postsináptica aparece un complejo de proteínas entrelazadas denominado densidad postsináptica. Las proteínas de la densidad postsináptica cumplen numerosas funciones, que van desde el anclaje y movimiento de receptores de neurotransmisores de la membrana plasmática, hasta el anclaje de varias proteínas reguladoras de la actividad de estos receptores.
Tipos de sinapsis]
Sinapsis eléctrica
Es aquella en la que la transmisión entre la primera neurona y la segunda no se produce por la secreción de un neurotransmisor, como en las sinapsis químicas (véase más abajo), sino por el paso de iones de una célula a otra a través de uniones gap, pequeños canales formados por el acoplamiento de complejos proteicos, basados en conexiones, en células estrechamente adheridas.
La sinapsis eléctrica es la más común en los vertebrados menos complejos y en algunos lugares del cerebro de los mamíferos. Son más rápidas que las sinapsis químicas pero menos plásticas; por lo demás, son menos propensas a alteraciones o modulación porque facilitan el intercambio entre los citoplasmas de iones y otras sustancias químicas. En los vertebrados son comunes en el corazón y el hígado.
Las sinapsis eléctricas tienen tres ventajas muy importantes:
La sinapsis eléctrica posee una transmisión bidireccional de los potenciales de acción, en cambio la sinapsis química solo posee la comunicación correccional.
En la sinapsis eléctrica hay una sincronización en la actividad neuronal, lo cual hace posible una acción coordinada entre ellas.
La comunicación es más rápida en la sinapsis eléctrica que en la química, debido a que los potenciales de acción pasan a través del canal proteico directamente sin necesidad de la liberación de los neurotransmisores.
Sinapsis química
La sinapsis química se establece entre células que están separadas entre sí por un espacio de unos 20-30 nanómetros (nm), la llamada hendidura sináptica.
La liberación de neurotransmisores es iniciada por la llegada de un impulso nervioso (o potencial de acción), y se produce mediante un proceso muy rápido de secreción celular: en el terminal nervioso presináptico, las vesículas que contienen los neurotransmisores permanecen ancladas y preparadas junto a la membrana sináptica. Cuando llega un potencial de acción se produce una entrada de iones calcio a través de los canales de calcio dependientes de voltaje. Los iones de calcio inician una cascada de reacciones que terminan haciendo que las membranas vesiculares se fusionen con la membrana presináptica y liberando su contenido a la hendidura sináptica. Los receptores del lado opuesto de la hendidura se unen a los neurotransmisores y fuerzan la apertura de los canales iónicos cercanos de la membrana postsináptica, haciendo que los iones fluyan hacia o desde el interior, cambiando el potencial de membrana local. El resultado es excitatorio en caso de flujos de despolarización, o inhibitorio en caso de flujos de hiperpolarización. El que una sinapsis sea excitatoria o inhibitoria depende del tipo o tipos de iones que se canalizan en los flujos postsinápticos, que a su vez es función del tipo de receptores y neurotransmisores que intervienen en la sinapsis.
La suma de los impulsos excitatorios e inhibitorios que llegan por todas las sinapsis que se relacionan con cada neurona (1000 a 200 000) determina si se produce o no la descarga del potencial de acción por el axón de esa neurona.
]
Se distinguen tres tipos principales de transmisión sináptica; los dos primeros mecanismos constituyen las fuerzas principales que rigen en los circuitos neuronales:
transmisión excitadora: aquella que incrementa la posibilidad de producir un potencial de acción;
transmisión inhibidora: aquella que reduce la posibilidad de producir un potencial de acción;
transmisión moduladora: aquella que cambia el patrón y/o la frecuencia de la actividad producida por las células involucradas.
.
Generalmente, si una sinapsis excitatoria es fuerte, un potencial de acción en la neurona presináptica iniciará otro potencial en la célula postsináptica. En una sinapsis débil, el potencial excitatorio postsináptico («PEPS») no alcanzará el umbral para la iniciación del potencial de acción. En el cerebro, cada neurona mantiene conexiones o sinapsis con muchas otras, pudiendo recibir cada una de ellas múltiples señales. Cuando se disparan potenciales de acción simultáneamente en varias neuronas que se unen en sinapsis débiles a otra neurona, pueden forzar el inicio de un impulso en esa célula a pesar de que las sinapsis son débiles.
Una neurona presináptica que libera neurotransmisores inhibitorios, como el GABA, puede generar un potencial inhibitorio postsináptico («PIPS») en la neurona postsináptica, bajando su sensibilidad y la probabilidad de que se genere un potencial de acción en ella. Así la respuesta de una neurona depende de las señales que recibe de otras, con las que puede tener distintos grados de influencia, dependiendo de la fuerza de la sinapsis con esa neurona. John Carew Eccles realizó algunos experimentos importantes en los inicios de la investigación sináptica, por los que recibió el Premio Nobel de Fisiología o Medicina en 1963.
Tras la fusión de las vesículas sinápticas y la liberación de las moléculas transmisoras en la hendidura sináptica, el neurotransmisor es rápidamente eliminado del espacio por proteínas especializadas en su reciclaje, situadas en las membranas tanto presináptica como postsináptica. Esta recaptación evita la desensibilización de los receptores postsinápticos y asegura que los potenciales de acción subsiguientes generen un PEP de la misma intensidad. La necesidad de una recaptación y el fenómeno de la desensibilización en los receptores y canales iónicos significa que la fuerza de la sinapsis puede disminuir si un tren de potenciales de acción llega en una sucesión rápida, un fenómeno que hace que exista una dependencia de la frecuencia en las sinapsis. El sistema nervioso se aprovecha de esta propiedad para computaciones, y puede ajustar las sinapsis mediante la fosforilación de las proteínas implicadas. El tamaño, número y tasa de reposición de las vesículas también está sujeto a regulación, así como otros muchos aspectos de la transmisión sináptica. Por ejemplo, un tipo de fármaco conocido como inhibidores selectivos de la recaptación de serotonina o SSRI afectan a ciertas sinapsis inhibiendo la recaptación del neurotransmisor serotonina. Por el contrario, un neurotransmisor excitatorio muy importante, la acetilcolina, no es recaptada, pero es eliminada por acción de la enzima acetilcolinesterasa.
La modificación de los parámetros sinápticos pueden modificar el comportamiento de los circuitos neurales y la interacción entre los diferentes módulos que componen el sistema nervioso (modal). Dichos cambios están englobados en un fenómeno conocido como neuroplasticidad o plasticidad neuronal.
El lenguaje químico del cerebro
Foto: Paweł Czerwiński en Unsplash.
A su vez, en el proceso actúan proteínas que hacen posible la sinapsis, conformando el proteoma sináptico.
Un nuevo estudio realizado por investigadores del Instituto de Ciencia y Tecnología de Okinawa, en Japón, descifra el lenguaje químico ligado al proteoma sináptico y revela su importancia en las redes cerebrales que favorecen la memoria, el aprendizaje, la atención o la ubicación espacial.
Según un artículo publicado en Medical Xpress, comprender este fascinante lenguaje molecular es de vital importancia por muchas razones, pero principalmente porque las fallas en el proceso sináptico forman parte de la raíz de una gran cantidad de enfermedades cerebrales, como el autismo, el Alzheimer, la epilepsia, el Parkinson o la esquizofrenia, entre otras.
La investigación de los especialistas japoneses, publicada en la revista Proceedings of the National Academy of Sciences (PNAS), podría favorecen nuevos abordajes y tratamientos al facilitar la comprensión del rol que juegan las proteínas en el circuito comunicacional del cerebro, como así también en su conexión con el resto del cuerpo.
Según el Dr. Zacharie Taoufiq, autor principal del estudio, “esta investigación ha dado como resultado un catálogo de todas las diferentes proteínas que participan en las sinapsis. Gracias a esta información contaremos con una gran base para estudiar la diversidad regional y evolutiva del cerebro a nivel sináptico. También será clave para encontrar la causa molecular de la enfermedad de cada paciente, una difícil tarea que nos espera en el futuro”, indicó.
Uno de los aspectos centrales de la investigación, en la que también participaron científicos del Instituto Max Planck de Química Biofísica en Göttingen, Alemania, y de la Universidad de Doshisha en Kioto, Japón, es el reconocimiento y caracterización de las llamadas vesículas sinápticas (SV). Se trata de complejos centros de procesamiento molecular y químico, que funcionan en el marco de una delicada interacción armónica para garantizar una correcta neurotransmisión.
Hasta el momento no se disponía de los datos relativos a la base molecular completa de las sinapsis, pero con la nueva investigación se contará ahora con el relevamiento más extenso y rico de las proteínas presentes en dichos procesos. Para llegar a estos resultados, los investigadores trabajaron en base a un método que les permitió descubrir muchas secuencias ocultas: el objetivo era identificar proteínas que pudieran parecerse en gran medida a otras, pero que presentaran funciones diferentes.
Los resultados superaron las expectativas de los científicos, ya que se hallaron 4.439 proteínas sinápticas, de las cuales 1.466 forman parte de vesículas sinápticas (SV), triplicando el catálogo existente en la actualidad. Al mismo tiempo, descubrieron una gran diversidad en las proteínas SV, que forman subpoblaciones con funciones muy concretas y específicas.
Todo indica que las proteínas implicadas en las sinapsis han desarrollado su propia estructura comunicacional. “Parece que los proteomas sinápticos están estructurados como verdaderos lenguajes, con unas pocas palabras (o proteínas) de uso frecuente y muchos términos menos habituales pero más específicos y significativos «, concluyó el Dr. Taoufiq.
La extensión del catálogo disponible de proteínas sinápticas tiene un valor que excede a su importancia científica, porque permitirá contar con una nueva herramienta para comprender el surgimiento de una gran cantidad de enfermedades cerebrales. Este conocimiento podrá desembocar en alternativas terapéuticas más eficaces, cuando por ejemplo en la actualidad los ensayos clínicos para el tratamiento del Alzheimer alcanzan una tasa de fracaso del 99,6%.
Bibliografía
Bear MF, Connors BW, Paradiso M.A: Neurociencia: explorando el cerebro. Barcelona: Masson, 2002. ISBN 84-458-1259-9.
Hormuzdi SG, Filippov MA, Mitropoulou G, Monyer H, Bruzzone R: «Electrical synapses: a dynamic signaling system that shapes the activity of neuronal networks». Biochim Biophys Acta. 2004 mar 23;1662(1-2):113-37. PMID 15033583.
Kandel ER, Schwartz JH, Jessell TM: Principios de neurociencia. Madrid: McGraw-Hill, 2001, 4.ª ed. ISBN 84-486-0311-7.
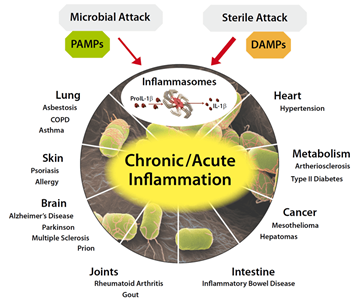
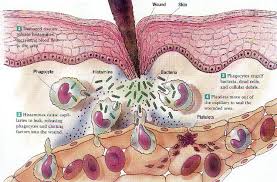
La inflamación es una respuesta del sistema inmune a un daño en el organismo. El daño puede ser causado por agentes de distinta naturaleza, por ejemplo mecánico (como puede ser un golpe o una fractura), infeccioso (por alguna bacteria o virus), químico (por contacto con alguna sustancia agresiva), etc.
El sistema inmune pone en marcha una serie de procesos necesarios para detectar, aislar y eliminar ese agente dañino. Posteriormente se iniciarán mecanismos de recuperación del tejido dañado. Estos procesos dan lugar a la característica tétrada de Celso: calor, rubor, tumor y dolor. Pero también y de manera dominanate. TRASTORNOS FUNCIONALES
Hay dos tipos de inflamacion
Aguda: de comienzo rápido y duración corta en la que predomina el exudado de fluido plasmático y la acumulación de linfocitos.
Crónica: que se produce cuando la inflamación aguda no se resuelve. Permanece en el tiempo, bien porque el patógeno no se pueda eliminar como el caso de infecciones latentes, por la persistencia de cuerpo extraño o porque se desarrolle un problema de autoinmunidad.
¿Cómo se convierte la inflamación en crónica?
En una situación de daño tisular se va produciendo un equilibrio entre infiltración celular, división, migración y muerte. En la inflamación aguda esa homeostasis evoluciona hacia la resolución y desaparición de la inflamación, sin embargo, en la inflamación crónica se produce una acumulación y activación persistente de células inmunes. En esta situación aumenta la secreción de citoquinas que prolonga la vida de los linfocitos y macrófagos dando lugar a la cronificación del proceso.
Pero desde los estudios que dignificaban a la inflamación, los estudios recientes, ven a esta como causante de la mayoría de las enfermedades degenerativas.
El trabajo de Andrew H. Miller 1 y Charles L. Raison 2 aclara parte de la alteraciones psiquiátricas en los procesos crónicos inflamatorios
La interferencia entre las vías inflamatorias y neurocircuitos en el cerebro puede conducir a respuestas conductuales, tales como evitación de la alarma, que probablemente se ha proporcionado una ventaja evolutiva en sus interacciones con patógenos y depredadores a los seres humanos tempranos. Sin embargo, en épocas modernas, estas interacciones entre la inflamación y el cerebro parecen impulsar el desarrollo de la depresión y pueden contribuir a la no respuesta a los tratamientos antidepresivos actuales. Datos recientes han aclarado los mecanismos por los cuales los sistemas inmunitarios innatos y adaptativos interactúan con neurotransmisores y neurocircuitos para influir en el riesgo para la depresión. Aquí detallamos nuestra comprensión actual de estas vías y discutir el potencial terapéutico de dirigirse el sistema inmunológico para tratar la depresión
Los conocimientos parciales llevan al error .
Quizás mas valida sea la frase del mal estudiante, tras un examen
“cuando sabia todas las respuestas, me cambiaron todas las preguntas”
La inflamación es mas que todo lo anterior, y los mecanismos reparadores de la inflamación son cada vez mas complejos
Sabemos desde hace muchos qué tiempo que la inflamación es un mecanismo de reparación . fue considerado siempre
Cuando nos parecía que conocíamos algo de la inflamación, aparecen múltiples trabajos de como se regula y cronifica la inflamación y la multiplicidad de metabolitos que intervienen
EL INFLAMASOMA es una estructura conformada por proteínas intracelulares implicadas en el inicio de la respuesta inflamatoria por estímulo intracelular..
El ensamblaje y la activación del inflamasoma es un proceso esencial en los mecanismos naturales de defensa inmune. El inflamasoma es una plataforma de multiproteínas citosólicas que permite la activación de las caspasas proinflamatorias, las cuales transforman el precursor de la interleukina-1beta (pro-IL-1beta) a la forma activa, lo que conduce a una poderosa respuesta inflamatoria.
Se ha demostrado que algunas patologías autoinmunes están provocadas por la proteína criopirina, producida por el gen CIAS1, y que pertenece a la familia de proteínas NOD-LRR, las cuales en principio, protegen a las células contra las infecciones microbiológicas.
El inflamasoma es un complejo multiproteico formado por la caspasa 1, PYCARD, una NALP y en ocasiones una caspasa 5 u 11. La composición exacta del inflamasoma depende del activador que inicia el ensamblaje del inflamasoma.
Un inflamasoma representa un complejo de alto peso molecular que activa las caspasas inflamatorias y activa las citocinas de la familia IL-1. Se han descrito varios inflamasomas y hasta ahora estaban definidos por la proteína NLR que contienen: el inflamasoma NLRP1 (NALP1), el inflamasoma NLRP3 (NALP3) y el inflamasoma IPAF (NLRC4).
Curiosamente, el inflamasoma AIM2, descrito recientemente, no contiene ningún miembro de la familia NLR. Los Iiflammasomas pueden activarse a través de múltiples señales, incluyendo bacterias, toxinas de origen microbiano, xenobióticos, PAMPs y DAMPs. Se sugiere que los dominios LRR de NLRP3 median la autorepresión, probablemente a través de las chaperonas SGT1 y HSP90 que mantienen NLRP3 en un estado inactivo. Al detectar sus respectivos ligandos, NLRP1 o NLRP3 se oligomerizan a través del dominio NACHT, lo que conduce a la agrupación de PYD y al reclutamiento de la proteína adaptadora ASC (proteína tipo speck asociada a apoptosis) debido a interacciones homotípicas PYD-PYD. En el caso de AIM2, la oligomerización es probablemente mediada por la agrupación en múltiples sitios de unión dentro de dsDNA y no por un dominio de oligomerización central como NACHT.
Las caspasas inflamatorias se acercan lo que permite la autoactivación y la formación de la caspasa activa. En el caso de la procaspasa-1, se forma un tetrámero p10 / p20 después de la autoescisión. Además de la caspasa-1, NLRP1 también puede reclutar caspasa-5 para el complejo, pero el papel de la caspasa-5 todavía está en debate. Al contrario de NLRP1, NLRP3 y AIM2, IPAF no recluta una molécula adaptadora, sino que interactúa directamente con la procaspasa-1 a través de su dominio CARD (consulte la Figura a continuación). Sin embargo, dependiendo del estímulo de IPAF, la activación máxima de caspasa-1 en sentido descendente de IPAF puede requerir ASC o NAIP. El ensamblaje de los diferentes inflamasomas provoca una cascada downstream. Sin embargo, la caspasa-1 parece ser la caspasa inflamatoria dominante asociada con los inflamasomas. Todas las caspasas inflamatorias tienen un dominio CARD seguido de un dominio que contiene el residuo catalítico cisteína y se denominan caspasas inflamatorias porque sus sustratos principales son citoquinas (como pro-IL-1β, pro-IL-18 y eventualmente pro-IL-33) Que se escinden a su forma activa y secretada. Además, la activación del inflamasoma puede llevar a la muerte de la célula huésped en ciertos tipos de células, conocida como piroptosis. Se cree que la piroptosis es importante para restringir la replicación intracelular de patógenos invasivos.
Aunque los mecanismos que regulan la actividad del inflamasoma siguen siendo poco conocidos, se identificaron varias proteínas que pueden interferir con la activación del inflamasoma y el procesamiento de la caspasa inflamatoria dependiente del inflamado. En general, se pueden distinguir dos subtipos de reguladores del inflamasoma: los que contienen un dominio CARD y los que tienen un dominio PYD. Dichas proteínas abarcan no solo los reguladores del inflamasoma derivados del huésped, sino también diversos factores de virulencia bacteriana que inhiben la activación de caspasa-1 y las proteínas virales PYD.
Como la IL-1β y otras citoquinas son actores clave en la respuesta inflamatoria, es tentador especular que la IL-1β, las caspasas inflamatorias y los inflamasomas desempeñan un papel importante en varias enfermedades. De hecho, algunas enfermedades hereditarias o adquiridas en humanos se han relacionado con una elevada IL-1β, algunas de las cuales pueden tratarse con antagonistas contra la IL-1β o su receptor. Varias enfermedades, conocidas como síndromos periódicos asociados a la criopirina (CAPS), se han relacionado directamente con mutaciones de NLRP3. Además, la gota, una enfermedad autoinflamatoria caracterizada por una inflamación severa de las articulaciones, está asociada con la deposición de cristales de MSU en las articulaciones, entre otras características. Como la MSU es un potente agonista del inflamasoma NLRP3, se cree que la IL-1β regulada por el inflamasoma ejerce un papel patógeno en la gota. Además, la secreción de IL-1β por el inflamasoma NLRP3 se desencadena por un alto nivel de glucosa extracelular en las células b. La IL-1β elevada es un factor de riesgo para el desarrollo de Diabetes Mellitus Tipo 2 (T2DM) y contribuye a la resistencia a la insulina. Esto funciona como un sensor para el estrés metabólico, como en la forma de urato monosódico (MSU) o hiperglucemia, la NLRP3 Es probable que el inflamasoma contribuya a la patogénesis de la gota o la DMT2, respectivamente. Además, varios reguladores del inflamasoma tienen una relevancia significativa en las enfermedades. En pacientes con FMF (fiebre mediterránea familiar), se demostró que la pirina estaba mutada. Los niveles elevados de IL-1β en la enfermedad autoinflamatoria PAPA (artritis piógena, pioderma gangrenoso y acné) se asocian con mutaciones en PSTPIP1, una proteína que interactúa con la pirina. En conjunto, esto indica la importancia de los reguladores de pirina e inflamasoma en las enfermedades autoinflamatorias y puede permitir nuevos puntos de entrada para el tratamiento de la enfermedad.
Como la IL-1β y otras citoquinas son actores clave en la respuesta inflamatoria, es obvio especular que la IL-1β, las caspasas inflamatorias y los inflamasomas desempeñan un papel importante en varias enfermedades (como muestra la imagen de la derecha). De hecho, algunas enfermedades hereditarias o adquiridas en humanos se han relacionado con niveles elevados de IL-1β. Varias enfermedades, conocidas como síndromes periódicos asociados a la criopirina (CAPS), se han relacionado directamente con mutaciones en el gen NLRP3. Esta enfermedad autoinflamatoria está caracterizada por producir una inflamación severa de las articulaciones, está asociada con la deposición de cristales de MSU en las articulaciones, entre otras características. Como la MSU es un potente agonista del inflamasoma NLRP3, se cree que la IL-1β regulada por el inflamasoma ejerce un papel patógeno en la gota. Además, la secreción de IL-1β por el inflamasoma NLRP3 se desencadena por un alto nivel de glucosa extracelular en las células b. La IL-1β elevada es un factor de riesgo para el desarrollo de Diabetes Mellitus Tipo 2 (T2DM) y contribuye a la resistencia a la insulina. Esto funciona como un sensor para el estrés metabólico, como en la forma de urato monosódico (MSU) o hiperglucemia, la NLRP3 Es probable que el inflamasoma contribuya a la patogénesis de la gota o la DMT2, respectivamente.
Además, varios reguladores del inflamasoma tienen una relevancia significativa en las enfermedades. En pacientes con FMF (fiebre mediterránea familiar), se demostró que la pirina estaba mutada. Los niveles elevados de IL-1β en la enfermedad autoinflamatoria PAPA (artritis piógena, pioderma gangrenoso y acné) se asocian con mutaciones en PSTPIP1, una proteína que interactúa con la pirina. En conjunto, esto indica la importancia de los reguladores de pirina e inflamasoma en las enfermedades autoinflamatorias y puede permitir nuevos puntos de entrada para el tratamiento de la enfermedad.
Los inflamasomas son complejos de múltiples proteínas implicados en la inflamación fisiológica y patológica. El inmonoblot para caspasa-1 es el método estándar para detectar la activación del inflamasoma, pero esta técnica presenta ciertas dificultades técnicas, en particular con la elección de protocolos de precipitación, separación y transferencia de proteínas. Los pasos de unión y activación también son cruciales para obtener un resultado de inmunoblot exitoso. A continuación se enumeran los Protocolos de Springer de la Serie de Métodos y Protocolos que utilizan los Anticuerpos Estándar que Adipogen para la detección de Inflammasomas.
No existe una sola enfermedad que no tenga un componente inflamatorio y con el riesgo de “Que se haga crónica”
El Deterioro Cerebral Tardio en la Hemorragia Subaracnoidea
Enrique Rubio; Isabel Porta, Jordi Vilalta y Josep Llorca
Aunque el deterioro cerebral tardio en la HSA es conocido desde hace mucho tiempo, su relación con el espamo de las grandes arterias cerebrales la hace Ecker y Reimenschneider en 1951 (3). Se trata de una disminución del calibre arterial mas intenso en la proximidad del aneurisma que se extiende por los vasos adyacentes y aparece en las primeras semanas despues de la HSA., pero no siempre el deterioro del paciente se acompaña de vasoespasmo, ni siempre el vasoespamos deteriorta al paciente. Este deterioro aparece entre el 3º y 9º dia despues de la HSA y en la angiografia se ve vasoespasmo entre el 30 y el 70% de los casos y solo es sintomatico entre el 20 y el 30%.
De una manera tradicional se le llama Vasoespasmo, (V). aunque este solo aparezca alguna vez
El V lesiona gravemente a los pacientes que lo sufren y el deficits permanente suele ocurrir entre 10% y el 20% y solo en el 7% la morbilidad es severa, siendo la mortalidad del 7% (7)
Con el uso del TC se observó que el vasoespasmo esta directamente relacionado con la cantidad de sangre acumulada en los espacios subaracnoideos (5,6).
Clinica y diagnostico del Deterioro Cerebral Tardio.
Se ha considerado que el V tiene dos expresiones no necesariamente coincidentes. Vasoespasmo Clinico y Vasoespasmo Radiografico (4).
El V. Clinico o V. sintomatico se debe a una isquemia focal que produce un deterioro focal o difuso en un un paciente que ha sufrido una HSA. Es mas frecuente en el territorio de la Cerebral anterior ( cuya clinica seria de indiferencia, tendencia al sueño descontrol de esfinteres, falta de motivacion y prension forzada ) que en la Cerebral media (clinica de hemiparesia y deficit del lenguaje ).
El V. radiografico es un estrechamiento arterial demostrado por angiografia, a menudo en forma de lentificacion del contraste y donde solo se rellenan correctamente las grandes arterias (3). Cuando el deficits neurologico se corresponde con el teritorio arterial espasmodizado visto en la angiografia se le conoce como V sintomatico
Una serie de procedimientos terapeuticos han permitido un mejor conociemiento de esta entidad.:
DTC: El aumento de la velocidad de circulación de la sangre arterial detectado por Doppler transcraneal. La diferencia de diametro en la arteria carotica interna y la cerebral media y alteración del pulso intracraneral (1,10,11). En este estudio solo se utilizara la velocidad media.
MCA Velocidad En cm
MCA-ICA Relación
Grado
< 120
<3
Normal
120-200
3-6
Moderado
> 200
>6
Severo
La medición del flujo sanguineo cerebral (CBF) con xenon, no esta al alcance de todos los hospitales y aunque puede medir cambios importantes en CBF, no detecta defectos focales del mismo. (8,12).
La medición del BCF con la tomografia de emisión de positrones (PET) es el metodo ideal pero tiene un costo inalcanzable para la mayoria de los hospitales. (9)
El SPECT, que mide solo el BFC y parte del metabolismo es un metodo mas al generalizado y tienen gran fiabilidad (2).
Material y metodo.-
En el año en curso 1999 se han estudiado 12 pacientes que deterioraron tras sufrir una HSA, bien espontaneamente o despues de procedimientos terapeuticos. A estos pacientes se le practicaron los siguientes estudios, que se sumaban a los estudios de rutina.
TC a su ingreso y repetirlo dependiendo de su evolución, la media es de 3 TC por enfermos
DTC diario o mas de una vez al dia
SPECT el dia que comenzaba su deterioro y en algunos casos otro cuando empezaba las condiciones lo permitian.
Un total de 12 enfermos se han deteriorado, y en su estudio consiste este trabajo
Resultados
Nº enfermo
Espas mo angiografico
Embolizado
Operado
TAC
TCD Velocidad
SPECT
GOS
Relacion con el deterioro
1
Si
Si
No
Edema
Normal
Hiperemia
1
Angio-Oclu
2
Si
No
No
Edema Hidroce Falia
Alta
Isquemia T
5
No
3
Si
No
No
Edema
Alta
Isquemia T
5
No
4
Si
No
Si
Infarto
Normal
Isquemia F
1
No
5
No
Si
No
Edema
Normal
Isquemia-Normal
1
No
6
No
Si
No
Infato
Normal
Isquemia Hemisferio
5
Angio-Oclu
7
No
Si
No
Infarto
Normal
Isquemia F Normalizacion
1
No
8
Si
No
Si
Infarto
Normal
Isquemia F Hiperemia
2
Cirugia
9
No
Si
No
Infarto
Normal
Isquemia T
5
NO HIV
10
Si
No
Si
Edema
Alta
Isquemia T
5
No
11
No
Si
No
Hemorragia talamo
Normal
Isquemia F. Diasquisis
2
No
12
No
No
Si
Hidrocefalia
5- Pacientes fallecieron
7..- Obtuvieron buenos resultados
De los 5 fallecidos.
3.- tenian vasoespasmo radiografico y velocidades de mas de 120 en el TCD. En los 3 se observo una severa isquemia difusa de todo el cerebro en el SPECT y edema difuso en el TAC.
1.- tenia vasoespasmo en la angiografia y las velocidades en la TCD eran normales, isquemia en el hemisferior izdo en el SPECT e infarto hemisferico en la TAC
1.- paciente HIV +, se deterioro espontaneamente y presento amplio infarto en la TAC, velocidades normales en el TCD e isquemia difusa aunque mas acentuada en un hemisferuio en el SPECT
De los que obtuvieron buenos resultados
Nº 1.- presentó vasoespasmo en la angiografia , TCD normal e hiperemia en la mitad posterior del craneo en el SPECT, edema e hidrocefalia en la TAC
Nº 4.- Vasoespasmo en la angiografia, TCD normal y SPECT lesion isquemica frontal derecha. Infarto en la TAC
Nº 5.- No vasoespasmo en la angio. TCD velocidades normales y en el SPECT, reducción del flujo cortical de manera transitoria y reversible y edema en la TAC.
Nº 7.- No vasoespasmo angiografiaco, TCD normal, SPECT lesion fisquemica frontal derecha que mejoro en un 2º SPECT.e infarto en el TAC.
Nº 8.- Tenia vasoespasmo angiografico, TCD normal y en el SPECT lesion isquemica frontal y posteriormente hiperemia cortical difusa e infarto en el TAC.
Nº 11.- No vasoespasmo angiografico, TCD normal.. Conservacion de la perfusion en el SPECT . Hermorragia talamica en la TC.
Nº 12.- No vasoespasmo en la angio. TCD normal y en el SPECT defecto temporal derecho . Hidrocefalia en el TAC
Causas del deterioro
En 7 casos no hubo una causa clara que explicara el deterioro
En 1 caso el paciente era HIV positivo
En 2 casos fue la angiografia-oclusion el desencadenante del deterioro, en uno de ellos aparecio espontaneamente una hemorragía talamica
En 2 casos fue la cirugía el desencadenante del deterioro
De los 5 casos que fallecieron , 3 se deterioraron espontaneamente. Uno en relación con el HIV + y otro a partir de la oclusión del aneurisma por via artertial.
En conclusion.
La lesión que secundariamente aparece en la HSA, es plural:
a).- El verdadero vasoespasmo ocurre en el 25% de los paciientes que deterioraron,se caracteriza por; vasoespasmo angiografico, aumento de la velocidad del TCD, isquemia difusa intensa en el SPECT y edema-necrosis amplio en el TAC,
b).- Los procedimientos terapeuticos, tales como la oclusión arterial del aneurisma, o el HIV+, son capaces de producir en pacientes que no tienen vasoespasmo angiografico y velocidades normales en la TCD, una lesion isquemica amplia en el TAC y el SPECT incompatibles con la vida del enfermo. 16,6%.
d).- En el 41,6%, el paciente deterioró por una lesión isquemica focal que es parcialmente reversible, no proporciona necesariamente malos resultados y en 2 de ello el deterioro empezó siguiendo tras procedimientos terapeuticos, 1 por oclusion arterial y 1 por oclusion quirurgica.
e).- En un 16,6% la lesion isquemica focal cursa con hiperemia . Los dos casos de hiperemia coinciden con la realización de procedimientos terapeuticos, oclusion por via arterial y clipaje del aneurisma por cirugía.
Se puede deducir de lo anterior, que en la HSA,:
El vasoespasmo tipico, clinico, angiografico y TCD, solo aparece en el 25%, y cursa con lesiones cerebrales muy amplias que suelen ser incompatibles con la vida.
Las lesiones isquemicas cerebrales amplias, sin vasoespasmo angiografico ni TCD, también son incompatbles con la vida, y pueden ser desencadenadas por procedimientos terapeuticos o enfermedades intercurrentes.
Las lesiones focales, sin vasoespasmo, y con isquemia focal en el SPECT y TAC, son soportables y suelen curar. En estos casos la hiperemia no parece ser un factor pronostico
BIBLIOGRAFIA
Cardoso ER, Reddy K. Bose D: Effect of Subarachnoid Hemorraghe on Intracranial Pulse Waves in Cats. J Nneurosurg 69:712-8, 1988.
Costa DC; Ell PJ: Brain Blood Flow in Neurology and Psychiatry. Series Editor:P.J.Ell,1992
Ecker A, Reimenschneider PA, Arteriographic demostration of spasm of the intracranial arteries. J Neurosurg 8:660-7,1951.
Ecker A, Reimenschneider PA, Arteriographic demostration of spasm of the intracranial arteries. J Neurosurg 1951:8:660.
Fisher C, Roberson GH, Ojeman RG. Cerebral vasoespasm with rupture of saccular aneurysm-the clinical manifestations. Neurosurgery 1977:1:245-248.
Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasoespasm to subarachnoid hemorrhage visualized by computarized tomographic scanning. Neurosurgery 1980;6:1-9.
Kassell NF, Sasaki T, Colohan AR, Nazar G: Cerebral vasoespasm following aneurysmal subarachnoid hemorrhage. Stroke 1985;16:562-572-. 39.
Knuckney N W, Fox K A, Surveyor I, et al.: Early Cerebral Blood Flow and CT in Predicting Ischemia after Cerehral Aneurysm Rupture J Neurosurg 62: 850-5. 1985.
Powers. W J, Grubb R L., Baker RP, et al.: Regional Cerebral Blood Flow and Metabolism in Reversihle Ischemia due to Vasospasm: Determination by Positron Emission Tomography. J NeurosurR 62:539-46, 1985.
Seiler RW. Grolimund P, Aaslid R et al: Cerebral vasospasm. Evaluated by Transcraneal Ultrasound Correlated with Clinical Grade and CT visualized Subarachnoid Hemorraghe. J Neurosurg 64:594-600, 1986.
Sekhar LN, Weschler LR, Yonas H. et al: Value of Transcranial Doppler Examination in the Diagnosis of cerebral Vasospasm after Subarachnoid Hemorraghe. Neurosurgery 22:813-21, 1988.
Weir B, Menon D, Overton T: Regional Cerehral Blood Flow in Patiens with Aneurysms: Estimation by Xenon 133 Inhalalion Can J Neurol SEi 5: 301-5. 1978.
Generalidades:- Los AI ocurren entre el 1 y el 8% de las autopsias generales y su expresión clinica mas frecuente es la hemorragia subaracnoidea (HS), la cual tiene una incidencia de 15/100.000. habitantes/año, y de ellas el 1 a 1.4 % son debidas a aneurismas. En USA las HS por AI matan o dejan incapacitados a 18.000 personas cada año (14)
Del estudio Cooperativo de Sahs y Drake en 1981 (1), se deduce; que entre el 8 y el 60% moriran antes de llegar al hospital y despues de llegar al hospital, moriran el 37%, quedaran severamente incapacitados el 17% y solo el 47% tendran un resultado favorable.
A pesar de los esfuerzos, la mortalidad de la HS no ha disminuido en los ultimos 30 años. Especial enfasis se ha puesto en evitar el resangrado de los AI, disminuir las complicaciones del vasoespasmo y disminuir las complicaciones quirurgicas, utilizando una terapeutica agresiva con los casos graves.
HEMORRAGIAS SUBARACNOIDEAS
Incidencia
Debidas a Aneurismas
Aneurismas en Autopsias
10 a 15/100.000 habitantes año
1 al 1.4%
1 al 8% de la poblacion general
Resultados de la Hemorragias Subaracnoideas
a los 3 meses de producida la hemorragia y segun el estudio cooperativo de 1981
Muertos antes de llegar al hospital
Muertos en el hospital
Quedan severamente incapacitados
Resultados favorables
Entre 8 y 60%
37%
17%
47%
ETIOPATOGENIA
La incidencia de AI depende de la edad y son lesiones adquiridas en su mayor cuantia. Se desarrollan en la bifurcacion de las arterias del poligono de Willis, donde es mas intenso el choque hemodinamico. Los desordenes geneticos ocurren en un pequeño porcentaje de ellos y entonces se ven AI familiares y en niños y se asocian a alteraciones del mesenquima tales como la Poliquistosis renal cuyo 10% sufren AI, Displasia fibromuscular, MAV cuyo 10% sufre AI, enfermedad de Moya Moya, ausencia unilateral de la carotida u oclusion de la misma, coartacion de la Aorta en el adulto, Ehlers-Danlos sindrome en su variedad tipo IV, sindrome de Marfan, Pseudoxantoma Elasticum, anemias sicke cell.
La alteracion del Colageno tipo III es la alteracion mas constante encontrada en los AI.
La mayor parte de los AI tienen un origen multifactorial,
1.- Defectos de la pared vascular, que puede a su vez ser consecuencia de un defecto genetico.
2.- Hipertension o strests hemodinamico.
3.- Arteriosclerosis que predispone especialmente al desarrollo de las ectasias arteriales.
4.- La gran longevidad del ser humano.
Las neoplasias, los traumatismos cranerales producen un tipo de AI, diferente que suelen localizarse en la periferia de las grandes arterias fuera del poligono de Willis. Las fracturas de la base del craneo pueden asociarse a aneurismas del segmento petroso de la carotica interna.
MANIFESTACIONES CLINICAS.-
Pueden ser sintomaticos y asintomaticos. Cuando son sintomaticos, la posibilidad de ruptura y de dar una HS es mayor que cuando no dan sintomas, y el sintoma es siempre un signo de aviso de una devastante hemorragia, asimismo la proporcion de ruptura de aneurisma es mayor cuando el aneurisma es sintomatico que cuado es asintomatico. En el estudio cooperativo (1) durante 5 años de evolución el 26% de los pacientes con aneurismas sintomaticos no rotos murio de HS en comparacion con solo el 2,6% de los que tenian aneurismas asintomaticos
La HS es la primera manifestacion clinica en el 90% de los casos (2), y el 80% de las HS no traumaticas estan producidas por la ruptura de un aneurisma. Se trata de un subito y fuerte dolor de cabeza que puede asociarse a nauseas y vomitos.
Los pacientes con HS severa derioran su estado de conciencia y la rapida hipertension endocraneal y la disminucion subita de su perfusion cerebral les conduce a parada respiratoria.
Entre el 40 y el 50% de los pacientes con HS por AI, han sufrido signos de alarma por HS minimas y tales signos suelen preceder unos 10 a 20 dias la ruptura del AI (3.4).
El grado de conciencia y los deficits neurologicos son factores que indican el pronostico de la HS, que son un buen indicador de la severidad del daño cerebral. El daño cerebral en la HS es causado por la isquemia que induce la hipertensión intracraneal inicial y no suele ser causado por el efecto masa, lo que explica que la evacuación del hematoma cuando existe, no suele acompañarse de mejoría del estado de conciencia.
Los aumentos de la PIC en la HS, inicialmente no son altos, y mejor no se mantienen, de forma que en la opinión del autor, el el brusco desplazamiento del tallo cerebro, condicionado por la salida a gran presión de sangre arterial al espacio subaaracnoideo. Esta perdida de conciencia que es muy frecuente es muy breve, y es similar a una conmoción cerebral
La gradacion de las HS se hace con distintas escalas, la de La Federacion Mundial de Neurocirujanos (WFNS)(5) Hunt & Hess (6) y Botterell(7). En un grupo de pacientes vistos en la Clinica Mayo (8), el 48% tenia el grado I y II de Hunt y Hess, , 20% tenian el grado III y 30% los grados IV y V. Estas proporciones varian de unos hospitales a otros dependiendo de donde le envian los pacientes (9)
Escalas en las Hemorragias Subaracnoideas
Modificada de Botterell
Grado
Descripcion
0
No Hemorragia o No rotura en los ultimos 30 dias
1
Con o sin moderadas cefaleas, alerta, orientado, no deficits motores o sensitivos
2
Severas cefaleas y signos meningeos mayores , con alteracion del sensorio o deficits focales
3
Mayor alteracion del sensorio o mayores deficits focales
4
Semicomatoso o comatoso , con o sin signos mayores de localizacion
Clasificacion de Hunt y Hess
Grados
Descripcion
0
No rotura
1
Asintomatico o minima cefaleas y ligera rigides de nuca
2
Modearada a severa cefaleas, rigides de nuca, no deficits neurologicos (aparte pares craneales)
3
Adormilado, confuso, deficits focales moderados
4
Estupor, moderada a severa hemiparesia, posible rigides de descerebracion inicial, y disturbios vegetativos
5
Coma profundo. rigides de descerebracion y apariencia de moribundo
Word Federation of Neurological Surgeons
Grado
Glasgow Coma Scale
Deficits Motor
1
15
Ausente
2
13 – 14
Ausente
3
13 – 14
Presente
4
7 – 12
Ausente o presente
5
3 – 6
Ausente o presente
El pronostico de las HS viene condicionado por el resangrado, el vasoespasmo y la hidrocefalia.
Escala de Fisher para las HS. segun la disposición de la sangre en la TC de las Hemorragias Subaracnoideas.
Grupo
Descripcion
1
No se detecto sangre
2
Deposito de una delgada capa de sangre con lamina vertical (interhemisferica, cisura Silviana,cister- na ambiens) menos de 1 mm de espesor.
3
Coagulos localiados en forma de hojas vertical de 1 mm de espesor (o ambos)
4
HS difusa o no pero con sangre intracerebral o intraventricular
El espasmo esta en relación con la cantida de sangre depositada en los espacios subaracnoideos. Los enfermos de grado 3 tienen mas alto riesgo (96%) para el desarrollo de espamos clinico, que los de otros grupos (10)
Los grados clinicos y de TC, junto al tamaño del AI y el estado del paciente, son usados para determinar; la indicación, el tiempo de operacion de los AI
Con frecuencia la HS se asocia a hematoma intraparenquimatoso e intraventricular, que pueden ser controladas por evacuacion del hematoma o drenaje ventricular y usando el activador del plasminogeno o irrigación con uroquinasa (11). El daño cerebral dependera de la magnitud del hematoma y en algunos casos el paciente se puede beneficiar de la evacuación del hematoma o del drenaje ventricular.
Efecto Masa de los AI. Este fenomeno ocurre en los AI en apoximadamente un 10% y suelen corresponder a AI grandes, aunque en ocaciones pequelños AI pueden ejercer presion sobre estructuras neurales adyacentes (12). Los sintomas suelen ser: Dolor retroorbitario, paralisis del II par, sindrome del seno cavernoso, hidrocefalia, deficits de campos visuales, henmiparesias leves y nmoderadas y disfuncion hipotalamo-pituitaria. En los casos de aneurismas de crecimiento rapido, el dolor se produce por la distencion que el saco aneurisma produce sobre la duramadre. El mayor problemas vienen condicionado por la posibilidad inmediata del ruptura del AI (13), por lo que la indicacion quirurgica es inmediata.
Complicaciones de la HS.
Resangrado
Hidrocefalia
Espasmo y deterioro cerebral por isquemia y deficits neurologicos
RESANGRADO
Es la complicacion mas frecuente y dramatica en los primeos momentos despues de la HS. El mayor riesgo de resangrado ocurre en las primeras 24 horas (4.1%) y en los restantes 13 dias va decreciendo a 1,4% cada dia. El riesgo acumulado de resangrado en los primeros 14 dias es del 19% y en los primeros 6 meses este riego es del 40 al 50% y posteriormente un 3% anual (15.16). La mortalidad debida al resangrado llega al 70% ( 17.18) y para evitar el resangrado se indica la cirugía en primer lugar.
HIDROCEFALIA
Ocurre precoz y tardiamente en la HS. En las primeras fases de la HS es una hidrocefalia oclusiva, pues los coagulos obstruyen la circulación del LCR en los ventriculos y en las cisternas (19). Ocurre entre el 10 y el 30% de los pacientes con HS y es demostrado por el CT , y en el 30 al 40% de aquellos pacientes es asintomatico (20.21). En los casos sintomaticos y con dilatacion ventricular en la TC, debe colocarse rapidamente un drenaje ventricular para evitar el deterioro neurologico, evitando el drenaje excesivo, para evitar que se desequiliobre la presion intramural del AI y se produsca el resangrado. En general los pacientes con hidrocfelaia tienen peor pronostico que aquellos con ventriculos no dilatados (20).
En fases mas tardias la hidrocefalia es producida por el bloqueo que hace la sangre en las granulaciones aracnoideas y la alteracion en la reabsorcion del LCR. Ocurre entre el 10 y el 15% de los casos (21.22). Se produce en tales casos un sindrome de la hidrocfelaia del adulto, con incontinencia de esfintres, deterioro de la marcha y de las funciones cognitivas y la CT muestra la tipica dilatacion ventricular, incluyendo el IV ventriculo y posiblemente la transependimal reabsorción del LCR. El tratamienrto de la hidrocfgelia en esta fase es la colocacion de un shunt ventricular permanente (22.24)
VASOESPASMO ARTERIAL CEREBRAL TARDIO
Se trata de un fenomeno que frecuentemente ocurre despues de la HS y en el que los grandes vasos arteriales cerebrales se espasmodizan entre los 3 y 9 dias despues de la HS (25.26) . A pesar de los cuantiosos estudios sobre el tema en los ultimos 30 años se ha avanzado poco en el conocimiento y tratamiento del vasospesmo tras la HS (27.28)
Los productos derivados de la lisis del coagulo en los espacios subaracnoideos, oxihemoglobina, leurotrienios, acido araquidonico son los causantes del vasoespasmo. Los deficits nutritivos de la pared arterial han sido considerados tambien como causantes del espasmo. El vasoespasmo esta directamente relacionado con la cantidad de sangre acumulada en los espacios subaracnoideos como demostró Fisher (10.29).
En la angiografia el vasoespasmo se ve entre el 30 y el 70% de los casos y solo es sintomatico entre el 20 y el 30%. La morbilidad por deficits permanente suele ocurrir en 10 al 20% y solo en el 7% la morbilidad es severa, siendo la mortalidad del 7% (30).
CLASIFICACION DE LOS AI.
Los AI se clasifican en razon de su etiologia, tamaño, localizacion y forma:
ETIOLOGIA
TAMAÑO, del Saco Aneurismatico
FORMA
Idiopaticos Anormalidades en la pared arterial influenciadas por factores congenitos y el stres hemodinamico. Se localizan en el poligono de Willis
Aneurisma SacularMenos de 1,5 cm
Sacular, que es el mas frecuente, y tienen un cuello bien demitido y un saco aunque el cuello esta a veces oculto por las arterias vecinas
Alteraciones especificas. Riñon poliquistico, coartacion de la aorta, Displasia fibromuscular Conectivopatias. MoyaMoya. Oclusion unilateral de carotida o vertebral. Agenesia congenita de arteria o persistencia circulacion embrionaria. Se localizan en el poligono de Willis
Aneurisma Globular de 1,5 a 2,5 cm
Fusiforme. Difusa dilataciono de una arteria sin cuello, son tipicos de la arterisoclerosis y se observan tambien en la aorta, y tambien son secundarios con frecuencia a infecciones, traumas y tumores
Factores acquiridos: Traumas. Tumores. Metastasis. Infecciosos. Se disponen en las arterias perifericas
Aneurisma Gigante, mas de 2,5 cm.
Disecantes de la pared de la arteria, son raros intracraneales y frecuentes en carotida y vertebral en el cuello y se manifiesta por isquemia y embolismos. Son mas frecuentes en la circulacion posterior
Falso. Generalmente debidos a truamas e infecciones y no existe verdadero aneurisma sino un tejido cicatrical reactivo, facilemnte rompible y dificil de disecar
EXAMEN DE LA HS.-
Es conocida la frecuencia con la que la HS altera las funcion cardiaca, por lo que todos los pacientes deben tener un ECG, monitorizacion de gaes en sangre y en aquellos pacientes con sospecha de presion venosa aumentada, se les debe medir la presión venosa central, y la presión cardiaca de salida a traves de un cateter de Swan-Ganz. (31.32)
Examen Neurologico
El examen neurologico a la entrada del paciente, seguido de examenes periodico es imprescindible para detectar el estado del paciente y sus futuros cambios de evolución, resangrado, hidrocefalias y espasmos y debe usarse algunas de las escalas de monitorizacion de loa AI (5.6.7). El examen precos del fondo de ojo diagnosticaria la hemorragia subretional (subhialoidea) que ocurre en el 10% de los paciente con HS y que necesita de un tratamiento especial otalmologico (33).
2.- Lindsay KW, Bone I, Callander R. Neurologya and Neurosurgery Ilustrated. 2nd ed. New York: Churchill Livinstone, 1991
3.- LeBlanc R. The minor leak preceding subaracchnoid hemorrhage. J Neurosur1987;66:35-39.
4.- Okawa SH. Warning signs prior to rupture of an intracranial aneurysms. J Neurosurg 1973: 38:578-580.
5.- Drake CG (chairman). Report of Word Federation of Neurosurgical Surgeon Committee on a Universal Subarachnoid Hemorrhage Grading Scale (letter). J Neurosurg 1988;68:985-986.
6.- Hunt W, Hess RM. Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J Neurosurg 1968:28:14-20.
7.- Botterell EH, Logheed WM, SCott JW, Vandewater SL. Hypothermia and interruption of carotid, or carotid and vertebral circulation, in the surgical management of intracranial aneurysms. J Neurosurg 1956; 13:1-42.
8.- Phillips LH II, Whisnant JP, O’Fallon WM, Sund TM Jr. The unchanging pattern of subarachnoid Hemorrhage in a community. Neurology 1980;30;1034-1040.
9.- Whisnant JP, Sacco SE, O’Fallon WM, Fode NC, Sundt TM Jr. Referral bias in aneurysmal subarachnoid hemorrhage. J Neurosurg 1993;78:726-732.
10.- Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasoespasm to subarachnoid hemorrhage visualized by computarized tomographic scanning. Neurosurgery 1980;6:1-9.
11.- Todo T, Usui M, Takakura K. Treatment of severe intraventriculoar hemorrhage by intraventricular infusion of urokinase. J Neurosurg 1991;74:81-86
13.- Sundt TM Jr. Surgical Techniques for Saccular and Giant Intracraneal Aneurysms. Baltimore: Willians & Wilkins, 1990
14.- Torner JC: Epidemiology of subarachnoid hemorrage. Semin Neurol 1984;4:354-369.
15.- Jane JA, Kassell NF, Torner JC, Winn HR. The natural history of aneurysm and arteriovenous malformation. J Neurosurg 1985;62:321-323.
16.- Kassell NF. Torner JC. Aneurysmal rebleeding a preliminary report from de Cooperative Aneurysmal Study. Neurosurgery 1983:13:479-481
17.- Lindsay KW, Bone J, Callander R. Neurology and Neurosurgery Ilustrated 2 nd e New York: Churchill Livingstone, 1991
18.- Nishiota H, Torner JC, Graf CJ, Kassell NF, Sahs AL, Goettler LC. Cooperative study of intracranial aneurysm and subarachoid hemorrhage : a long term pronostic study. II Rupture intracranial aneurysms managed conservatively. Arch Neurol 1984;41:1142-1146
19.- Graff-Radford NR, Torner J, Adams HP Jr, Kassell NF. Factors associated with hydrocephalus after subarachnoid hemorrhage: a report of the Cooperative Aneurysms Study. Archi Neurol 1989;46:744-752.
20.- van Gijn J, Hijdra A, Wijdicks EPM, Vermeulen M, van Crevel H. Acute Hydrocephalus after subarachnoid hemorrhage. J Neurosurg 1985;63:355-362.
21.- Vassilouthis J, Richardson AE, Ventricular dilatation and communicantin hydrocephalus following spontyaneous subachnoid hemorrhage. J Neurosurg 1979;51:341-351.
22.- Vassilouthis J, The syndrome of normal-plexus hydrocephalus. J Neurosurg 1984;61:501-509.
23.- Hayashi M, KObayashi H, Munemoto S, Nosaky J, Higashi S, Yamamoto S, et al. An analysis of time course of intracranial pressure in patients with comumunicatin hydrocephalus following subarachnoid hemorrhage due to rupture intracranial aneurysm. Brain Nerve 1982;34:653-660
24.- Black PM, Ojeman RG, Tzouras A, CSF s unt for dementia, incontinence and gait disturbance. Clin Neurosur 1985;32:632-651.
25.- Ecker A, Riemenschneider PA, Arteriogrphy demostration of spasm of the intracranial erteries : with special reference to sacular arterial aneurysm. J Neurosurg 1951;8:660-667
26.- Weir B, Grace M, Hansen J, Rohberg C: Time course of vasoespasm in man. J Neurosur 1978;48:173-178.
27.- Kassell NF, Sasaki T, Colohan AR, Nazar G: Cerebral vosoespasm following aneurysmal subarachnoid hemorrhage. Stroke 1985;16:562-572.
28.- Wilkins RH: Attented prevention of treatment of intracranial arterial spasm: a survey. Neurosurgery 1980;6:198-210.
29.- Fisher C, Roberson GH, Ojeman RG. Cerebral vasoespasm with rupture of saccular aneurysm-the clinical manifestations. Neurosurgery 1977:1:245-248.
30.- Kassell NF, Sasaki T, Colohan AR, Nazar G: Cerebral vasoespasm following aneurysmal subarachnoid hemorrhage. Stroke 1985;16:562-572.
31.- Crompton MR. Hypothalamic lesion following the rupture of the cerebral berry aneurysms. Brain 1963; 83:301-314.
33.- Garfinkle AM. Danys JR. Nicolle DA. Colohan AR. Brem S. Terson’s syndrome a reversible cause of blindness following subarachnoid hemorrhage. J Neurosurg 1992;76:766-771.
Según datos de la Organización Mundial de la Salud (OMS), cada tres segundos se diagnostica un nuevo caso de demencia en el mundo. Tres de cada cinco de esos diagnósticos serán de la enfermedad de Alzheimer, el tipo más común de demencia. La dificultad para encontrar un fármaco que detenga su declive cognitivo (más de 200 posibles tratamientos han fracasado en los ensayos clínicos en pacientes) ha provocado que uno de los principales retos de la investigación en estos momentos se centre en la prevención y en la detección precoz.
Sobre esos retos y sobre el grandísimo impacto del Alzheimer en las familias de los afectados debatieron la doctora Mercè Boada, neuróloga y fundadora y directora médica de Fundación ACE, y el doctor Arcadi Navarro, catedrático de Genética, profesor de Investigación ICREA en la Universidad Pompeu Fabra de Barcelona y director del Barcelona Brain Research Center (BBRC) y de la Fundación Pasqual Maragall, en un encuentro digital enmarcado dentro del ciclo Debates de Vanguardia, organizado por Fundación La Caixa.
En un mundo con la esperanza de vida en aumento, la ausencia de un tratamiento para prevenir o detener el curso del Alzheimer puede triplicar el número de casos en 2050, lo que en 30 años podría hacernos hablar ya de una epidemia. “Estamos delante de una bomba de relojería”, aseguró la doctora Mercè Boada, que destacó que hablamos de una patología “socialmente agresiva” (por su impacto en cuidadores, sistemas de salud y en la economía) y difícilmente soportable para el sistema.
«El Alzheimer es una enfermedad socialmente agresiva y difícilmente soportable para el sistema”
Su opinión la compartió Arcadi Navarro, que comparó el Alzheimer con la actual pandemia provocada por la covid-19. “Estamos viviendo una pandemia gravísima, pero circunstancial. Ya vemos la luz al final del túnel. Con el Alzheimer, sin embargo, no pasa eso. Hablamos de una pandemia estructural, una ola que avanza lentamente, que no llama tanto la atención y en la que los poderes públicos no están invirtiendo suficiente ni en investigación ni en cuidados de las personas que sufren la enfermedad”.
Ambos expertos destacaron el impacto económico “brutal” que tiene la enfermedad y que asciende cada año a decenas de miles de millones de euros, un gasto que, además, recae mayoritariamente sobre las familias de los afectados. “La diferencia con otras enfermedades como el cáncer es que en éstas el coste lo asume la sociedad a través del Sistema de Salud. En el Alzheimer, sin embargo, el 80% del coste lo asume la familia, lo que supone una causa de precarización tremenda”, denunció Navarro, que recordó que han llevado al Congreso de los Diputados una propuesta para el cambio del modelo de atención a los afectados por una enfermedad “proporcionalmente muy mal atendida” y cuyo peso suele recaer sobre los hombros de las mujeres. “Según un estudio que realizamos, el 65% de los cuidadores eran mujeres; por este orden: la esposa del afectado, la hija y la esposa del hijo mayor. También vimos que había muchos pacientes leves diagnosticados de demencia que vivían solos. ¿Quiénes eran mayoritariamente? Mujeres”, añadió Boada.
Tanto Mercè Boada como Arcadi Navarro se mostraron en todo momento optimistas con el futuro de la enfermedad.
En primera instancia, por el aumento de la conciencia de la población y de los médicos, lo que ha facilitado que aumente el número de pacientes que llegan a centros especializados como la Fundación ACE en un estado aún leve de la enfermedad, cuando hace no tanto, lo normal era que lo hiciesen en una fase moderada-grave.
El Alzheimer es una pandemia estructural que avanza lentamente y no llama la atención social”
En segundo lugar, por el desarrollo de más y mejores técnicas para el diagnóstico precoz de los pacientes que hace un lustro ni siquiera existían. “Tenemos algoritmos de Inteligencia Artificial que son capaces de clasificar ante una imagen del cerebro si éste ya muestra un síntoma de deterioro aunque no se haya manifestado, hay biomarcadores en sangre que clasifican con una certeza del 90% la acumulación de proteínas…”, enumeró Navarro, que aseguró que estos avances les hacen ser optimistas, ya que ante un diagnóstico precoz existe la opción de prescribir tratamientos no farmacológicos para aumentar la resiliencia cerebral.
En ese sentido, Mercè Boada destacó la importancia de que poco a poco estas técnicas de diagnóstico precoz del Alzheimer se integren en nuestro ADN como hoy en día, por ejemplo, está integrada la importancia de hacerse una mamografía para detectar el cáncer de mama en fases tempranas de su desarrollo.
Los expertos, por último, se mostraron optimistas respecto al desarrollo de fármacos que en un futuro próximo permitan llegar a un “escenario ideal” en el que cada paciente de Alzheimer, como ocurre en el caso del cáncer, tenga “un tratamiento a medida con una multiplicidad de alternativas terapéuticas”.
Querida Mercedes siempre te he admirado por tú afición y constancia en el estudio de las enfermedades neurodegenerativas. Me parece deducir de tu trabajo que no eres , o si le eres, lo es escasamene, optimista en su pronóstico y me asusta la idea de una epidemia terrible, para la que no tenemos ninguna solución efectiva y que cada día se extiende más.
Yo estoy obsesionado con la idea de la inflamación. las proteínas de depósito que tienen las enfermedades degenerativas, son macrofagos que se depositan sobre algo qué está debajo. muy probablemente gérmenes y varios.
Me llama la atención, que tú que te lo lees todo no hables de los gérmenes como primer causante de estas enfermedades concretamente del Alzheimer. Ya sabes qué Eloy Alzheimer , hasta que no se descubrió la proteína beta amiloide como depósito, creía que era una enfermedad infecciosa, como hizo después Rover Moir , que acaba de morir víctima de un Glioblastoma. Y cómo sigue reclamando el doctor Carrasco microbiólogo de la Universidad de Madrid desde hace 20 años , que reclama el haber descubierto repetidas veces varios tipos de gérmenes, sobre todo hongos en el cerebro de estos enfermos, sobre todo con ELA.
La fotografía que circula, en la que se ven las proteínas depositándose sobre gérmenes es
Gérmenes en el cerebro procedentes del intestino
El patrón de toda enfermedad neurodegenerativa es el mismo , Un macrófago que segrega una proteína que se deposita es una estructura nerviosa vital . y yo estoy ilusionado, con que esta proteína tapa a una serie de gérmenes que están debajo y qué son la fuente que desencadena todo lo demás.
Amiloides envolviendo Levaduras
Prácticamente toda la medicación para la enfermedad de Alzheimer intenta eliminar la proteínas beta amiloide o la Tau, Y hasta ahora y que yo sepa, no lo han conseguido. La apuesta de los laboratorios por conseguir una medicación efectiva en esta enfermedad es enorme.
Luis Carrasco es un científico que desde hace unos 20 años insiste en la posibilidad de que las enfermedades neurodegenerativas tengan como origen un germen o varios a la vez . No es el primero que afirma esto, ya Rober Moir, insistio y lucho por demostrar este postulado. Y a su muerte, sigue sin admitirse la teoría microbiana, que a mi me parece indiscutible. La aparicion reciente de dos casos de Parkison, por el coronavirus, potencian esta idea, de que cualquier germen puede asentar en el cerebro, lesionarlo y desencadenar una respuesta inmunitaria, que es la que perpetua el daño.
Si lleganos a la conclusion, que si, que son germenes, o varios a la vez, los antibioticos, las vacunas, o lo que toque, podria evitar estas epidemias de enfermedades cronicas y mutilantes.
Mientras tanto, solo tenemos mas de lo mismo
Gracias Mercedes por tu entusiasmo y perdona mi atrevimiento
En noviembre de 1906, en la XXXVII Conferencia de Psiquiatría del Sudoeste Alemán, Alzheimer presentó la comunicación «Sobre una enfermedad específica de la corteza cerebral», en la que se hacía por primera vez la descripción de una inusual enfermedad de la corteza cerebral cuyos síntomas principales eran pérdida de memoria, desorientación, alucinaciones y finalmente muerte. La enfermedad se había diagnosticado en Augusta D., una mujer de 51 años que ingresó en 1901 en el Hospital de Frankfurt con un cuadro clínico cuyas manifestaciones eran delirio de celos, pérdida de memoria, alucinaciones, desorientación temporo espacial, paranoia, alteraciones de conducta y un grave trastorno del lenguaje. Finalmente, Augusta falleció por una infección a causa de las escaras producto de la inmovilidad e infección pulmonar.
Cuando el Dr. Alzheimer estudió el cerebro de Augusta, encontró que la corteza cerebral era más estrecha de lo normal (atrófica) y, además, que existían dos tipos de anomalías muy llamativas, que hoy en día siguen siendo las características histopatológicas principales de la Enfermedad de Alzheimer:
Placas, especie de esferas o acúmulos extraneuronales constituidos por un material anómalo (denominado, desde un principio, amiloide por teñirse como el almidón, pero que hoy se sabe que es de naturaleza proteica) y que puede contener terminaciones de neuronas degeneradas y estar rodeado de células gliales reactivas.
Ovillos neurofibrilares, acumulaciones de material aparentemente fibroso en el interior de las propias neuronas.
Alois Alzheimer pensó la demencia de aquella paciente que tanto le impresionó era debida a un germen y esto se mantuvo en general hasta que se encontró un depósito de proteína beta amiloidótica, a partir de entonces existe una opinión muy extendida, que esta proteína junto a la TAU, se precipitan sobre determinadas estructuras del cerebro y son las responsables de la enfermedad de Alzheimer
Las fibras amiloides, son la forma anómala de la proteína responsable de la homeostasis del ioncalcio intracelular, induce la apoptosis celular. Se sabe también que la Aβ se acumula selectivamente en las mitocondrias de las células cerebrales afectadas en el alzhéimer y que es capaz de inhibir ciertas funciones enzimáticas, así como alterar la utilización de la glucosa por las neuronas.
Desde hace mucho tiempo se interpretó pero el depósito de estas proteínas anormales como un fenómeno inflamatorio y la intervención de las citoquinas pueden también jugar un papel en la patología de la enfermedad de Alzheimer.
La inflamación es el marcador general de daño en los tejidos en cualquier enfermedad y puede ser el daño producido en el alzhéimer por la expresión de una respuesta inmunológica.
Existe evidencia de que, Amiloide, ApoE4 y envejecimiento, aumentan el riesgo de la enfermedad de Alzheimer. Pero existen dudas, de la incidencia y aparición de cada uno de estos factores sobre si cada factor debilita la memoria en el estado cognitivamente saludable.
Los Australian Imaging, Biomarkers and Lifestyle (AIBL) En un análisis durante seis años, vieron que la memoria episódica disminuye solo en los ancianos con depositos de amiloide cerebral.
Y concluyeron que:
ApoE4 cambia la pérdida de memoria relacionada con amiloide una década antes.
Sin amiloide cerebral, los portadores de ApoE4 envejecen sin pérdida de memoria.
En el 22 de enero JAMA Neurology, los investigadores dirigidos por Paul Maruff, Universidad de Melbourne, Australia, informan que en personas sin amiloide cerebral, la memoria episódica desciende por debajo de un umbral de normalidad a partir de los 76 años, mientras que que en portadores de ApoE con amiloide cerebral, esto comienza una década más temprano. Sorprendentemente, sin embargo, las personas sin amiloide cerebral recuerdan muy bien, incluso aquellos que portan un alelo ApoE4. «Esto desafía la noción de pérdida de memoria relacionada con la edad, ya que se asume ampliamente que el envejecimiento en sí mismo juega un papel en el declive». Esto lo afirma , el primer autor, Yen Ying Lim, a Alzforum. En cambio, parece que Aβ explica gran parte de la variación en la disminución de la memoria observada en los estudios de envejecimiento, dijo Lim.
Este estudio se suma otros que están comenzando a mostrar que la función cognitiva permanece relativamente estable a lo largo del tiempo cuando no hay amiloide ( Lim et al., 2016 ; Jack et al., 2015).
Para descubrir cómo interactúan el amiloide, la edad y la ApoE, Lim y sus colegas incluyeron 447 participantes cognitivamente normales de AIBL en su análisis. Tienen un promedio de 72.5 años. Los científicos los genotiparon para ApoE, tomaron sus valores de PET amiloide cerebral al inicio, luego dividieron la cohorte en cuatro grupos dependiendo del estado de Aβ y ApoE4. En promedio, el grupo amiloide positivo fue cuatro años mayor que el grupo amiloide negativo. Todos los participantes tomaron pruebas de memoria episódica cada 18 meses durante seis años, y los investigadores combinaron los resultados de la prueba de aprendizaje verbal de California, la prueba de memoria retardada de memoria lógica y la prueba de figura compleja en una puntuación compuesta.
Los positivos de amiloide fueron los únicos con pérdida de memoria episódica. Su declive comenzó, en promedio, alrededor de los 76 años. Si un participante también portaba un alelo ApoE4, su declive comenzó casi una década antes: a los 65 años. A los 85 años, los portadores de ApoE4 positivos a amiloide habían caído 1.5 unidades de desviación estándar por debajo de amiloide -negativos no portadores en el compuesto, característicos de alguien que tiene problemas para recordar los recuerdos adquiridos recientemente y se acerca al deterioro cognitivo leve, dijo Lim. Los no portadores de amiloide positivo habían disminuido en 0,7. Por el contrario, la memoria episódica se mantuvo estable en personas sin placas, independientemente de su estado ApoE4.
Los autores utilizaron un modelo cuadrático transversal para correlacionar la edad con el cambio en la memoria episódica . Esto explica mejor la varianza que los modelos lineales que se habían utilizado en la mayoría de los otros estudios. El modelo sugiere que la disminución de la memoria en las personas positivas para Aβ puede acelerarse con la edad avanzada, y que esta aceleración puede deberse a ApoE4.
Los datos confirman lo que los estudios más cortos insinuaron, a saber, que ApoE4 por sí solo no aumenta el riesgo de pérdida de memoria en personas cognitivamente sanas ( Mormino et al., 2014 ).
«La única contribicion del estudio AIBL es el largo seguimiento clínico de los participantes, que fortalece la confianza en las conclusiones sobre las relaciones entre amiloide, ApoE y deterioro cognitivo», afirma Clifford Jack de la Clínica Mayo en Rochester. La sospecha apareció después de analizar datos de estudios longitudinales en la Clínica Mayo.
«El mecanismo principal por el cual ApoE4 afecta negativamente la cognición está mediado por la amiloidosis», afirma Alzforum.
Es importante demostrar esta interacción entre ApoE4 y amiloide. Ya anteriormente se había pensado que la memoria tiene un impacto en portadores de ApoE4 cognitivamente saludables ( Caselli et al., 2009 ). Sin embargo, ese estudio no determinó si los sujetos tenían amiloide en el cerebro, por lo que no pudo decir si ApoE4 funcionaba solo o a través de amiloide.
Con fines terapéuticos , los ensayos de prevención que están basados en el genotipo ApoE, nos recuerdan que una intervención dirigida al amiloide debe aplicarse a aquellos individuos cuyos cerebros albergan amiloide, y que no podemos suponer que lo hacen en base a la posesión de un E4 alelo «,
No se menciona en este trabajo, como ApoE4 afecta la acumulación del amiloide en este estudio. Se afirma, que si un portador de ApoE4 es amiloide negativo, entonces su memoria no disminuirá durante al menos seis años, según este estudio. Estas decisiones pueden ser útiles para determinar el riesgo de disminución de la memoria asociada con Aβ y ApoE4 en cada edad.
Los autores del trabajo, advierten que, dado que la muestra proviene de una población seleccionada, mayoritariamente con educación universitaria, excluyendo a las personas con factores de riesgo cardiovascular y otras afecciones médicas y psiquiátricas, es necesario repetirla en una muestra basada en la población. También advierten que tenían muy pocos homocigotos de ApoE4 para saber qué hacen dos copias de este alelo a lo largo del tiempo. Finalmente, dado que solo utilizaron escaneos iniciales para este análisis, los autores no calcularon cómo la disminución de la memoria se correlacionaba con la cantidad de acumulación, o cómo ApoE4 afectaba esa acumulación.
Existe algunos trabajos, que están de acuerdo en el deposito anormal, de proteínas en el Alzheimer y otras enfermedades degenerativas, pero son los resultados de un fenómeno inflamatorio de respuesta a la invasión de uno o varios gérmenes .
Esto lo mantiene de manera sostenida el profesor Carrasco y Moir, y tienen una lógica aplastante.
Primero un germen, después un macrófago segrega una proteína para destruirlos, y cuando estas proteínas superan, ciertos niveles, aparece la enfermedad.
Pese a lo bondadosa que parece esta teoría, no tienen todavía mucha aceptación.
La enfermedad de Parkinson puede formar parte de un síndrome extrapiramidal producido por un germen sobre el que se deposita una proteína llamada cuerpos de Levy . Y lesiona preferentemente las zonas dopaminérgicas. Algunos actores están afirmando, qué tras la epidemia de coronavirus que estamos padeciendo, otra epidemia en pacientes crónicos de tipo extrapiramidal aparecerá y de hecho ya hay algunos casos descritos. Y ello no es extraño qué varios parénquimas estén afectados al mismo tiempo. Los artículos que vienen a continuación , podrían explicar este postulado
A menudo, las personas afectadas por la enfermedad de Parkinson comienzan teniendo síntomas que difieren mucho de un paciente a otro, una circunstancia que ha suscitado numerosas especulaciones. Una reciente investigación ha apuntado la existencia de dos subtipos de la enfermedad, según el lugar del organismo en el que comience: el cerebro o el sistema nervioso autónomo periférico. Mediante tomografía por emisión de positrones e imágenes de resonancia magnética, los investigadores examinaron a 37 pacientes con enfermedad de Parkinson de novo. También se incluyó en el estudio a 22 personas que aún no habían sido diagnosticadas pero tenían un alto riesgo de desarrollar la enfermedad. Los resultados del estudio demuestran que algunos pacientes tenían dañado el sistema dopaminérgico del cerebro antes de que se produjeran lesiones en el sistema nervioso autónomo periférico. En otros pacientes, las pruebas revelaron daños en el sistema nervioso autónomo periférico (intestinales y cardíacos) antes de que el daño en el sistema dopaminérgico cerebral fuera visible. Y ALGUNOS pacientes con enfermedad de Parkinson tienen un microbioma intestinal distinto al de las personas sanas, podría ser particularmente interesante, en el caso de la enfermedad de Parkinson iniciada fuera del cerebro, estudiar el microbioma intestinal del paciente porque podría ser factible frenar la patología mediante un tratamiento dirigido al microbioma.
¿Que se afecta primero en la enfermedad de Parkinson, el cerebro o el cuerpo ¿
La enfermedad de Parkinson se caracteriza por la presencia de agregados de α-sinucleína intraneuronales anormales, que pueden propagarse de una célula a otra de forma priónica. Sin embargo, sigue siendo incierto dónde se originan los agregados iniciales de α-sinucleína. Hemos planteado la hipótesis de que la enfermedad de Parkinson comprende dos subtipos. Un tipo de cerebro primero (de arriba hacia abajo), donde la patología de la α-sinucleína surge inicialmente en el cerebro con una extensión secundaria al sistema nervioso autónomo periférico; y un tipo de cuerpo primero (de abajo hacia arriba), donde la patología se origina en el sistema nervioso autónomo entérico o periférico y luego se disemina al cerebro. También planteamos la hipótesis de que el trastorno de conducta del sueño REM aislado (iRBD) es un fenotipo prodrómico para el tipo de cuerpo primero. Usando imágenes multimodales, Probamos la hipótesis cuantificando la disfunción neuronal en estructuras correspondientes a la afectación de las etapas I, II y III de Braak en tres grupos distintos de pacientes. Incluimos 37 consecutivos pacientes de novo con enfermedad de Parkinson en este estudio PET de casos y controles. Los pacientes con enfermedad de Parkinson se dividieron en 24 casos RBD negativos (PD RBD− ) y 13 casos RBD positivos (PD RBD + ) y un grupo comparador de 22 pacientes iRBD. Usamos 11 C-donepezil PET / CT para evaluar la inervación colinérgica (parasimpática), gammagrafía con 123 I-metayodobencilguanidina (MIBG) para medir la inervación simpática cardíaca, resonancia magnética sensible a neuromelanina para medir la integridad de las neuronas pigmentadas del locus coeruleus y 18PET con F-dihidroxifenilalanina (FDOPA) para evaluar la capacidad de almacenamiento de dopamina putaminal. El volumen del colon y los tiempos de tránsito se evaluaron con tomografías computarizadas y marcadores radiopacos. Los datos de imágenes de los tres grupos se interrogaron con ANOVA y pruebas de Kruskal-Wallis corregidas para múltiples comparaciones.
Los PD RBD y PD RBD + grupos mostraron marcadas reducciones similares en la captación de putaminal FDOPA-específico, mientras que dos tercios de los pacientes tenían IRBD exploraciones normales ( P < 10 -13 , ANOVA). Cuando se compara con los PD RBD pacientes, los PD RBD + pacientes y IRBD demostraron una reducción cardíaca media MIBG: relaciones de mediastino ( P < 10 -5 , ANOVA) y el colon11 Valores de captación estándar de C-donepezilo ( P = 0,008, ANOVA). El grupo PD RBD + tendió a una reducción media del locus coeruleus: pons de la resonancia magnética en comparación con PD RBD- ( P = 0,07, prueba t ).
En comparación con los otros grupos, el grupo PD RBD + también tenía volúmenes de colon agrandados ( P < 0,001, ANOVA) y tiempos de tránsito colónico retrasados ( P = 0,01, Kruskal-Wallis). Los datos del paciente combinados de iRBD y PD RBD + fueron compatibles con una trayectoria de cuerpo primero, caracterizada por la pérdida inicial de la señal MIBG cardíaca y 11Señal de donepezilo C-colónico seguida de pérdida de captación de FDOPA putaminal. Por el contrario, los datos de PD RBD− fueron compatibles con una trayectoria de cerebro primero, caracterizada por una pérdida primaria de la captación de FDOPA putaminal seguida de una pérdida secundaria de la señal de MIBG cardíaca y de la señal de 11 C-donepezil. Estos hallazgos apoyan la existencia de subtipos de la enfermedad de Parkinson
Esta investigación me parece buena, pero los gérmenes, seguro que van por donde pueden y en poco tiempo
Si asientan de forman preferente, lo hacen en las grandes cavidades y de preferencia en el intestino y cuando las condiciones les son adversas y aumenta la porosidad intestinal, se va a donde un antígeno receptor se lo permita.
Primero el cerebro y después el cuerpo. O todo a la vez