| EL GEN MYC
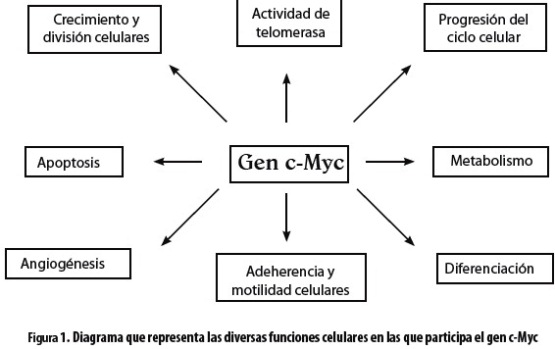
La familia de genes MYC es una de las más ampliamente estudiadas (1). Dichos genes actúan como factores de transcripción y reguladores del ciclo celular e intervienen en la proliferación, apoptosis y diferenciación celular y en la inmortalización (2). Los genes c-Myc, N-Myc and L-Myc se expresan en diferentes tejidos durante la embriogénesis. Específicamente, el gen c-Myc se expresa principalmente en células con una mayor tasa de proliferación. Por su parte, el N-Myc se expresa a bajos niveles en diversos tejidos neonatales y especialmente en células pre-B, riñón, cerebro e intestino (3, 4). Por otra parte, el L-Myc se expresa durante el desarrollo del riñón y el pulmón y en los compartimentos de diferenciación y proliferación del cerebro y del tubo neural. Los tres genes participan activamente en los mecanismos de señalización celular. Además, sintetizan factores de transcripción que forman heterodímeros con la proteína Max, para luego unirse al ADN; de esta manera, regulan la expresión de múltiples genes (1,5,6). El gen más ampliamente estudiado de esta familia es el c-Myc, que fue el primero en ser descubierto mediante su homología con el gen transformante del virus de la mielocitomatosis aviar MC29 (v-Myc) (7). Los otros dos genes, N-Myc y L-Myc, se descubrieron posteriormente por su homología con el v-Myc en secuencias amplificadas en células de neuroblastoma y en tumores de células pequeñas del pulmón, respectivamente (2,8-10) En 1911 Peyton Rous observó que el sarcoma del pollo podría transmitirse mediante extractos libres de células de tumores y sugirió que un virus podría ser el agente etiológico de los sarcomas (11,12). Con base en el trabajo de Rous, Bishop y colaboradores (13) llevaron a cabo un estudio en un subgrupo específico de retrovirus aviares, que incluía diversos tumores en pollos, como leucemia mieloide, tumores de hígado, riñón y sarcomas; estos estudios condujeron a la identificación del oncogén v-Myc en pollos. Luego, con el descubrimiento de un gen homólogo, denominado c-Myc en pollos, se corroboró la hipótesis de que los retrovirus aviares oncogénicos podrían interaccionar con genes reguladores del crecimiento celular y transmitir el gen activado. Por otra parte, se encontró que el gen c-Myc se encuentra alterado en diversos tumores sólidos, leucemias y linfomas, así como en diferentes tumores en animales (2,14-16). En las células neoplásicas es muy frecuente que se presente la amplificación del gen c-Myc en tumores hematopoyéticos debido a translocaciones cromosómicas o aneuploidías. Estas alteraciones inducen una desregulación de la expresión del gen. Se sabe que en el locus del gen c-Myc (8q24) ocurren frecuentemente reordenamientos cromosómicos, además de integración de virus oncogénicos que promueven modificaciones funcionales o estructurales (17,18). Entre las alteraciones cromosómicas más comunes que involucran al locus del c-Myc está la translocación t(8;14) presente en el linfoma de Burkitt (LB) (18-20). Así, el gen c-Myc translocado actúa de forma defectuosa. De otro lado, la amplificación del gen c-Myc es muy frecuente en el cáncer de mama, pulmón, ovario, próstata, leucemias y linfomas; mientras que la pérdida de la regulación es más común en el cáncer de colon, tumores ginecológicos y melanoma (1,18,20). Estructura y función del gen c-Myc El proto-oncogén c-Myc participa en una amplia red de vías metabólicas, y por esta razón tiene múltiples funciones como son la progresión del ciclo celular, metabolismo celular, angiogénesis, adherencia celular, reparación del ADN, apoptosis y diferenciación celulares (1) (figura 1). El gen c-Myc fue descubierto hace más de 25 años como un oncogén retroviral aviar (v-Myc) en células infectadas por el virus de transformación aguda MC29, el cual induce mielocitomatosis y tumores en pollos; posteriormente se identificó el c-Myc en humanos y en otros vertebrados por su homología celular con el v-Myc (1,13,21). El c-Myc se localiza en la región cromosómica 8q24. Codifica para una proteína con función de factor de transcripción nuclear, que interviene en numerosos mecanismos celulares y vías de transducción. En células normales el c-Myc se encuentra bien regulado, en contraste con la desregulación que presenta en las células cancerosas (9,19-22). El gen c-Myc se compone de tres exones. El exón 1 tiene dos promotores y no codifica. Los exones 2 y 3 codifican para la proteína Myc que tiene un sitio de inicio para la traducción en el nucleótido 16 del exón 2 (20); en dicho sitio se sintetiza una proteína c-Myc de 64 kDa constituida por 439 aminoácidos y presenta una traducción alternativa que origina dos tipos de proteínas, una de secuencia larga y otra de secuencia corta, denominadas p67 Myc (67 kDa) y MycS (45 kDa). Otros autores también informan un polipéptido de c-Myc de 45-kDa. De otro lado, se tiene un interés particular en las diferentes funciones de dos productos alternativos del gen c-Myc: c-Myc 1 y c-Myc 2. La proteína c-Myc 2 estimula el crecimiento celular, mientras que c-Myc 1 lo suprime; o sea, que las actividades de estas dos proteínas son antagónicas (23). Esta situación, sumada a la ocurrencia de mutaciones en el gen c-Myc en neoplasias humanas, hace más complejo el estudio del papel de este gen en dichas células. Por otra parte, se sabe que la pérdida de la regulación de la expresión del gen c-Myc ocurre por diferentes mecanismos genéticos, tales como la amplificación, alteraciones cromosómicas numéricas y estructurales, inserción viral o mutaciones puntuales; estas alteraciones se relacionan con la transformación e inmortalización de las células neoplásicas (1,24). Como resultado de múltiples estudios sobre la regulación de la expresión del gen c-Myc, se ha informado la existencia de una gran variedad de factores de transcripción que interaccionan con él. El gen c-Myc se regula por mecanismos complejos de transcripción y postranscripción. Se conocen cuatro promotores de transcripción, pero el ARN sintetizado a partir del promotor P2 contribuye con 80% a 90% del total del ARN en las células normales (25,26). En las células humanas normales el gen c-Myc se transcribe en diferentes tejidos, mientras que en células neoplásicas la transcripción se lleva a cabo de forma anormal (22). La actividad oncogénica del c-Myc se demostró en animales transgénicos y en estudios in vitro con cultivos celulares (27). Asimismo, se encontró que una deleción homocigótica del gen c-Myc es letal en embriones murinos, lo que sugiere que la expresión de este gen es esencial para el desarrollo embrionario (28). Estructura y función de la proteína c-Myc Los protooncogenes codifican para proteínas reguladoras de la transcripción, que son fundamentales para muchos procesos celulares. Estas proteínas, cuando se expresan de una forma anormal, actúan como oncoproteínas por lo que se asocian con el desarrollo de diversas neoplasias (16,29-31). La proteína c-Myc es una fosfoproteína nuclear de 439 aminoácidos que juega un papel importante en la regulación de la expresión génica en células humanas; generalmente forma complejos de cremallera de leucina (LZ, por la sigla en inglés de leucine zipper) con otras moléculas (8,9). Además, tiene varias secuencias estructurales conservadas y posee dos dominios principales. El primero es un motivo de dimerización, denominado helix-loop-helix leucine zipper (HLH/LZ) en el residuo C-terminal que consta de 90 aminoácidos; este dominio se requiere para la dimerización con otras proteínas y con el ADN (28,32). Además, el dominio HLH/LZ permite la dimerización homotípica o heterotípica con otras proteínas HLH/ LZ, como ocurre con la heterodimerización con la proteína Max y con la unión al ADN en una secuencia específica E-box, denominada sitio Myc E-box o EMS (por la sigla en inglés de E-box Myc Site); la alteración de este dominio destruye la actividad biológica de la proteína, indicando que la unión al ADN es esencial para su función (33). El segundo dominio comprende una gran parte de la proteína c-Myc y se define como el dominio de transactivación. La región N-terminal de c-Myc tiene 140 aminoácidos y contiene grupos ácidos ricos en prolina y glutamina, similares a los asociados con algunos dominios de transactivación (28, 33-35); la secuencia de la proteína c-Myc posee varios dominios N-terminales conservados, que se denominan cajas Myc, y se encuentran en proteínas homólogas relacionadas, como N-Myc y L-Myc. La región rica en glutamina de c-Myc es esencial para la actividad oncogénica. Además, el dominio N-terminal, denominado dominio de activación transcripcional (TAD) (aminoácidos 1 a 143) contiene un elemento rico en prolina que se extiende desde el aminoácido 41 hasta el 103 (36). El dominio TAD es necesario para activar la transcripción de c-Myc y para la transformación celular, la inhibición de la diferenciación celular y la inducción de la apoptosis mediada por c-Myc (28,37). De otro lado, en 1991 se identificó la proteína Max que interactúa con la proteína c-Myc para formar un heterodímero Myc-Max, complejo que se une luego al ADN (38). La proteína c- Myc tiene una vida media corta, de aproximadamente 20 minutos, mientras que la de la proteína Max es mayor de 24 horas; por lo tanto, en muchos sistemas, la proteína c-Myc es el componente limitante del heterodímero, lo cual es clave en la regulación de la transcripción génica en diversos mecanismos como la proliferación y diferenciación celulares y la apoptosis (5,6,33). La dimerización de las proteínas Myc y Max mediante los dominios HLH/LZ es importante para la unión de este complejo con el ADN en secuencias específicas de hexanucleótidos, denominadas cajas E (E boxes) (5′-CA[C/T]GTG-3′) (33). Por esta vía, c-Myc activa la transcripción de promotores que contienen la secuencia CACGTG (39). El heterodímero Myc-Max unido al ADN, interacciona a través de la región N-terminal de Myc con una variedad de proteínas involucradas en la transcripción de múltiples genes; entre estas proteínas se incluyen las TRRAP (por la sigla en inglés de transactivation-transformation domain-associated proteins), que se asocian con la histona acetilasa GCN5 (40). La acetilación de histonas puede luego marcar la cromatina para permitir el acceso de factores de transcripción que pertenecen a la maquinaria de aquellos de tipo general (5). Por otra parte, en estudios in vitro se encontró que la proteína c-Myc es inactiva cuando se encuentra sola, no forma homodímeros, ni se une al ADN; por lo tanto, requiere la dimerización con Max para unirse al ADN (27,39). En contraste, la proteína Max forma fácilmente homodímeros y se une directamente al ADN para inducir la transcripción. Otro aspecto importante es que la proteína Max puede unirse a otras proteínas como las de la familia Mad, para inducir represión de la transcripción (6,33). Los niveles de la proteína Mad, que son opuestos a los de c-Myc, aumentan durante la diferenciación celular, mientras que una baja expresión de la proteína Mad2 (Mxi-1) se asocia con el desarrollo de tumores en modelos murinos. El complejo Mad-Max, al contrario de Myc-Max, interacciona con las histonas deacetilasas que inducen estructuras compactas de la cromatina, lo cual limita el acceso de los factores de transcripción al ADN (33). Finalmente, se presentan modificaciones postraduccionales de la proteína c-Myc, como glicosilación y fosforilación, que alteran su vida media; por ejemplo, la fosforilación del dominio de transactivación transcripcional de c-Myc constituye un sustrato para la acción de factores de crecimiento regulados por las proteínas MAP quinasas (por la sigla en inglés de mitogenactivated protein kinases), así como para proteínas quinasas dependientes del ciclo celular. Este dominio tiene gran importancia porque se lo considera un blanco directo para la regulación del ciclo celular y de diferentes vías de señalización (figura 2). De otro lado, se ha observado que la inhibición de la proteína c-Myc podría bloquear vías de señalización mitógenas específicas y de esta manera facilitar la diferenciación celular (34,36,41). Función del c-Myc en la regulación del ciclo celular El gen c-Myc participa junto con muchos otros genes en la regulación del ciclo celular. Sin embargo, no se conoce bien su función en la activación de las redes metabólicas citosólica y mitocondrial durante la entrada de la célula al ciclo celular (42); c-Myc no solamente promueve el paso de las células de G0 a G1, sino que también durante toda la fase G1 del ciclo celular induce la transcripción de genes e interviene en el crecimiento y la proliferación celulares y en la apoptosis. Los estudios sugieren que c-Myc tiene la capacidad de activar la maquinaria del ciclo celular y se lo considera un gen »maestro» para la activación de muchos genes por diferentes vías metabólicas. Se sabe que este gen es clave para la activación de la glucólisis, mediante la regulación de genes que intervienen en ella con el fin de proporcionar energía durante todo el ciclo celular (21,43). En células en reposo la proteína c-Myc estimula el inicio de la mitosis, lo que sugiere que es una proteína esencial para el crecimiento celular continuo; además, es necesaria para varias fases del ciclo celular. No solo c-Myc es indispensable en el punto de transición G0/G1 de dicho ciclo, sino que también, como resultado de su activación, permite a las células salir de la fase G0 y continuar con la progresión del ciclo (42,44-47). Con respecto a las funciones del c-Myc en el ciclo celular, varios estudios determinaron, mediante la recombinación homóloga, que la inactivación de los dos alelos de este gen produce una prolongación significativa de la fase S (28,47). Asi mismo, se demostró en células Myc-nulas, es decir, que no expresan L-Myc ni N-Myc, una mayor prolongación de las fases G1 y G2, mientras que la fase S transcurre en un tiempo normal, por lo que se concluye que c-Myc es esencial durante las fases G1 y G2 del ciclo celular. Por otra parte, está bien definido que proteínas como las ciclinas, las quinasas dependientes de ciclinas (CDK, por la sigla en inglés de cycline-dependent kinases), inhibidores de las CDK (CKI, por la sigla en inglés de cycline kinases inhibitors) y otras proteínas reguladoras del ciclo celular son importantes para el funcionamiento de c-Myc. Una de las vías por las cuales c-Myc participa en la progresión del ciclo celular es la regulación de los genes de las ciclinas; por ejemplo, una expresión desregulada de c-Myc se asocia con un aumento en la expresión de las ciclinas A y E (42, 44,45). Además, en cuanto a la regulación de la fase G1, la interacción entre c-Myc y ciclina D1 es compleja y depende de diferentes estímulos. Además, c-Myc aumenta la expresión de las CDK por varios mecanismos, por ejemplo: coopera con la proteína RAS (por la sigla en inglés de RA-t sarcoma) para inducir el promotor de CDC2 (CDK1). Otra relación directa entre c-Myc y el ciclo celular es la capacidad de activar directamente genes como cdc25A y el de la ciclina E durante la progresión de dicho ciclo (42,45). El gen cdc25A codifica para una proteína fosfatasa que a su vez activa a CDK2 y CDK4. Además, la expresión de c-Myc disminuye el nivel del inhibidor p27 durante la regulación del ciclo celular en el punto G1/S; sin embargo, no se conoce con claridad el mecanismo por el cual c-Myc interfiere con la actividad del p27 (48). En la década de los años 90 Cleveland y colaboradores encontraron en estudios con líneas mieloides de células progenitoras que la desregulación del gen c-Myc induce el mecanismo de apoptosis (28,49); estas células dependen de la interleucina-3 (IL-3) para su crecimiento y para la expresión de c-Myc. En ausencia de IL-3, la expresión anormal de c-Myc conduce a las células hacia la fase S, para luego activar por esta vía la apoptosis y detener el ciclo celular; además, se observó que c-Myc afecta la transcripción de diferentes genes que intervienen en la apoptosis, como es el caso de TP53. c-Myc en la oncogénesis El proto-oncogén c-Myc es uno de los genes más comúnmente relacionados con el origen de una gran variedad de neoplasias humanas (14,34,44). La pérdida de regulación de c-Myc juega un papel importante en el origen del cáncer. Estudios con animales transgénicos demostraron que la desregulación de c-Myc es el principal evento que podría explicar la carcinogénesis en la mayoría de los tejidos (16), además de que induce a la transformación celular en modelos in vitro e in vivo (44). De los anteriores hallazgos, también se concluye que la sobrexpresión de c-Myc se encuentra en más del 50% de las neoplasias humanas y se asocia con un mal pronóstico y un fenotipo invasor. En el locus 8q24 del gen c-Myc ocurren con frecuencia alteraciones cromosómicas que afectan su estructura y función (18,22). Entre las alteraciones más comunes de c-Myc que lo relacionan con la oncogénesis está la translocación recíproca t(8;14) en individuos con linfoma de Burkitt, una malignidad hematológica de células B, caracterizada por ser muy agresiva y con un alto grado de proliferación (19,20). En este tipo de tumor c-Myc se transloca con uno de los genes de la cadena pesada de las inmunoglobulinas (IGH, por la sigla en inglés de immunoglobulin heavy chain) localizado en el cromosoma 14 (18,19,22,50). Estos dos genes translocados se activan de una forma anormal en las células afectadas, lo que conduce a una expresión del gen c-Myc constitutiva y desregulada; así se alcanzan niveles altos de expresión del producto del gen. Además, el gen IGH potencia la desregulación de c-Myc. La t(8;14) se observa en cerca del 85% de los casos de linfoma de Burkitt (50-52); sin embargo, dicha translocación no es exclusiva de este linfoma sino que también se la ha encontrado en otras neoplasias humanas, como es el caso de leucemias y del mieloma múltiple (19,53). Por otra parte, en otros estudios se encontró que en el linfoma de Burkitt también puede ocurrir que el gen c-Myc se transloque con otros genes de las inmunoglobulinas, generando translocaciones cromosómicas variantes como la t(2;8) y la t(8;22), como resultado de los diferentes sitios de ruptura cromosómica (18,54). De los anteriores hallazgos se concluye que el gen c-Myc juega un papel importante en la patogénesis del linfoma de Burkitt (19,55). Finalmente, otras alteraciones frecuentes en el locus 8q24 que afectan la expresión del gen c-Myc son deleciones, aneuploidías del cromosoma 8, amplificación, mutaciones puntuales e inserción viral (15,18,24,53); todas estas alteraciones se presentan en diversas neoplasias como en los cánceres de mama, pulmón, ovario, próstata, colon y estómago, así como en leucemias y linfomas (18,56-59). Inestabilidad genética en cáncer inducida por c-Myc El cáncer es un proceso evolutivo en el que las células normales adquieren un fenotipo maligno a partir de la acumulación de diversas alteraciones genéticas y epigenéticas que afectan a protooncogenes, genes supresores de tumores y genes de reparación del ADN (19,60-63). La inestabilidad genética (IG) es una característica propia de las células tumorales; este fenómeno favorece la aparición de aneuploidías y, además, genera un aumento en la tasa de mutaciones (64). En la IG se identifican dos tipos: la inestabilidad microsatelital (MSI, por la sigla en inglés de microsatellite instability), también conocida como MIN, en la que se presenta expansión o contracción del número de repeticiones de oligonucleótidos presentes en secuencias de microsatélites de genes de reparación, como es el caso de los genes MLH1 y MSH2 en individuos con cáncer colorrectal hereditario sin poliposis (63,64); el otro tipo es la inestabilidad cromosómica (CIN, por la sigla en inglés de chromosomal instability), que se refiere a las alteraciones cromosómicas numéricas y estructurales presentes en las células neoplásicas (63,65-67). Se considera que cerca del 50% de los tumores sólidos tienen alteraciones cromosómicas. Los dos tipos de inestabilidad se presentan en una amplia variedad de neoplasias. Cabe mencionar que entre los tipos de IG en el cáncer también se propone la inestabilidad telomérica, que a su vez promueve la inestabilidad cromosómica (63,65,68); este tipo de inestabilidad afecta la integridad del telómero y se la ha observado en varias neoplasias (66). La relación del gen c-Myc con la IG en las células tumorales se podría explicar por sus funciones en la proliferación y regulación del ciclo celular, pero más específicamente por la inducción de especies reactivas de oxígeno y la promoción de CIN, especialmente aneuploidías y tetraploidías (64,68). Por consiguiente, la desregulación de c-Myc afecta su interacción con otros genes responsables de la integridad del genoma, como son los genes supresores de tumores y los de la reparación del ADN, lo que ocasiona que se alteren diversos mecanismos celulares y genéticos; de esta forma se induce la aparición de un fenotipo mutador en las células neoplásicas. De lo anterior se concluye que el gen c-Myc actúa como un gen »maestro» que coordina la expresión de múltiples genes (1,18,42,64). Además, c-Myc también contribuye a la oncogénesis mediante la inducción de la IG por alteraciones específicas en el punto de control G1/S del ciclo celular, en el cual se presenta la respuesta al daño del ADN; en consecuencia, se acumulan diversos tipos de daños en la célula (42,64). Amplificación del gen c-Myc en la oncogénesis La amplificación es una de las alteraciones más comunes del oncogén c-Myc en diversas neoplasias humanas (30,69,70). Muchos estudios demuestran que la amplificación y la sobrexpresión del oncogén c-Myc son claves en la iniciación y progresión del cáncer, tal como se informa en leucemias, linfomas y tumores sólidos. Además, la amplificación se relaciona con la sobrexpresión de oncogenes, produciendo una ventaja selectiva y mayor tasa de proliferación a las células transformadas (60-62). El oncogén c-Myc se encuentra expresado en altos niveles en diversas neoplasias como las de mama, próstata, pulmón, colon y linfomas, y en la mayoría se asocia con mal pronóstico de la enfermedad (14,71,72); por ejemplo, las frecuencias de amplificación y sobrexpresión de c-Myc en el cáncer de mama varían ampliamente entre 1% y 94% y de 22% a 95%, respectivamente (60,61,73-76). En otro estudio en el que se analizaron cerca de 1.000 casos de cáncer de mama se encontró que la amplificación de c-Myc fue del 17%; además se halló que esta alteración se correlaciona con un mal pronóstico de la enfermedad (77,78). La amplificación de c-Myc también se ha observado en un 29% de los casos de cáncer de próstata y en un 40% de los de cáncer gástrico (57,58). La amplificación es una alteración genética que conduce al aumento en el número de copias de un gen o de grupos de genes contiguos, lo que ocasiona una expresión génica anormal (18,62). Entre las causas cromosómicas que originan la amplificación del oncogén c-Myc están las translocaciones, trisomías, duplicaciones e isocromosomas que involucren el locus 8q24 (51,68). Asimismo, dichas alteraciones afectan las vías de regulación de c-Myc sobre otros genes, tales como TP53 y RB, por lo que se propone que la amplificación de este gen se asocia con la inestabilidad genómica, la cual a su vez promueve la oncogénesis (14,66,78). Un tipo de amplificación son los cromosomas dobles diminutos (DM) (double minutes), que son pequeños fragmentos extracromosómicos que contienen genes amplificados, están presentes en múltiples copias y son frecuentes en diversos tumores sólidos y linfomas (17-19,53); los cromosomas DM generalmente se detectan con técnicas de citogenética convencional o molecular (FISH, por la sigla en inglés de fluorescent in situ hybridization). Otro tipo de amplificación son las denominadas regiones homogéneamente coloreadas (HSR, por la sigla en inglés de homogeneous staining regions) presentes en determinadas regiones cromosómicas, como en la del locus 8q24 del gen c-Myc; las HSR son un producto del conjunto de genes amplificados; estas regiones contienen varios cientos de genes amplificados y tienen un patrón de bandeo cromosómico anormal (17). Estos dos tipos de amplificación son comunes durante el desarrollo y la progresión del cáncer y afectan por lo general el número de copias de proto-oncogenes, las cuales alteran la tasa de proliferación celular, lo que promueve la inestabilidad genómica y, a su vez, el proceso de carcinogénesis en diversas neoplasias (18,64). Se puede detectar la amplificación de oncogenes en muestras de tumores humanos con las técnicas de hibridación in situ fluorescente (FISH) e hibridación genómica comparativa (CGH, por la sigla en inglés de comparative genomic hybridization), que son muy específicas y sensibles para ese propósito (53,60,79). En numerosos estudios con estas técnicas han hallado niveles altos de amplificación de muchos genes en células neoplásicas, especialmente con los genes c-Myc y HER-2 en tumores primarios de mama. Se considera que la co-amplificación de estos dos genes se relaciona con la presencia de otros tipos de alteraciones genéticas, al igual que con tumores más agresivos y del mal pronóstico (60,62,80). Por lo anterior, la amplificación de los genes c-Myc y HER-2 en el cáncer de mama se considera como un marcador molecular recurrente con valor pronóstico para las pacientes (60,62,81,82). Además, los hallazgos sugieren que la amplificación de c-Myc podría ocurrir en las etapas iniciales del desarrollo del cáncer de mama y simultáneamente presentarse mutaciones en el gen TP53. Estos conocimientos son de gran importancia para la biología del cáncer, porque han permitido el desarrollo de nuevas drogas antineoplásicas que inhiban la amplificación génica en determinadas neoplasias; el caso más conocido y de gran utilidad en oncología ha sido el del trastuzumab (Herceptin®), una droga diseñada para bloquear la amplificación del gen ERBB2 durante el tratamiento del cáncer de mama; el estudio de la amplificación de este gen es importante porque se considera un marcador de recurrencia de la enfermedad (62,80,83). En un estudio hecho por el Grupo de Genética Médica, evaluando la amplificación del gen c-Myc en muestras de cáncer de mama incluidas en bloques de parafina con la técnica FISH (datos sin publicar), se encontró una alta frecuencia de aneuploidía del cromosoma 8 y de amplificación del gen c-Myc, corroborando lo informado en la literatura que ambas alteraciones son comunes en este tipo de cáncer (figura 3). En los estudios con FISH es posible detectar la heterogeneidad genética intratumoral que se presenta durante el desarrollo del cáncer, lo que es una ventaja de la técnica FISH con respecto a otras técnicas moleculares. Finalmente, la caracterización molecular de las alteraciones del gen c-Myc continúa siendo un tema de gran interés en la genética del cáncer, debido a la asociación de estas alteraciones con la génesis de muchas neoplasias en humanos; los nuevos conocimientos aportarán información valiosa para desarrollar nuevas estrategias terapéuticas con el fin de controlar eficazmente la expresión del gen c-Myc en el desarrollo del cáncer. Un equipo de científicos ha verificado que inhibir la proteína Myc, implicada en el desarrollo de diversos tumores, es una estrategia terapéutica eficaz también contra el tumor cerebral más frecuente y de peor pronóstico, el glioma. El equipo ha confirmado con modelos preclínicos que la inhibición de Myc impide que las células tumorales se dividan y proliferen eficientemente. La inhibición preclínica de Myc se ha validado en ratones como estrategia terapéutica contra el astrocitoma. Este mismo grupo consiguió anteriormente eliminar tumores pulmonares en ratones transgénicos gracias a la misma estrategia, que consiste en expresar el transgén Omomyc en modelos de ratón. Confirmaron, además, que no aparecían efectos secundarios tras administrar tratamientos repetidos y a largo plazo y demostraron que no aparecía resistencia. Ahora, la inhibición preclínica de Myc se ha validado también como estrategia terapéutica contra el astrocitoma, un tipo de glioma, en modelos de ratón in vivo y en las células progenitoras de estos tumores in vitro. En estos animales, el tratamiento con el transgén Omomyc reduce drásticamente los tumores y mejora los síntomas asociados hasta que el ratón se recupera y actúa con total normalidad. Los ratones tratados con Omomyc sobrevivieron, mientras que no lo hicieron los ratones no tratados. El impacto terapéutico de Omomyc radica en su estructura, homóloga a la de Myc, que permite el bloqueo de la transcripción de los genes controlados por esta proteína. La inhibición de Myc provoca ‘defectos’ de las células tumorales y a menudo su muerte por inducción de aberraciones mitóticas, es decir, la imposibilidad de dividirse con normalidad.
BIBLIOGRÁFICA 1. Eilers M, Eisenman RN. Myc’s broad reach. Genes Dev. 2008 Oct 15;22(20):2755-66. 2. Gustafson WC, Weiss WA. Myc proteins as therapeutic targets. Oncogene. 2010 Mar 4;29(9):1249-59. 3. Chu D-K, Zhang J, Shi H, Dong G-L, Liu X-P, Wang W-Z. [Expression of candidate tumor suppressor gene N-Myc downstream-regulated gene 2 in colon cancer]. Zhonghua Wei Chang Wai Ke Za Zhi. 2008 Jul;11(4):354-7. 4. Knoepfler PS, Kenney AM. Neural precursor cycling at sonic speed: N-Myc pedals, GSK-3 brakes. Cell Cycle. 2006 Jan;5(1):47-52. [ Links ] 5. Hurlin PJ, Huang J. The MAX-interacting transcription factor network. Semin Cancer Biol. 2006 Aug;16(4):265-74. [ Links ] 6. Rottmann S, Lüscher B. The Mad side of the Max network: antagonizing the function of Myc and more. Curr Top Microbiol Immunol. 2006 Jan;302:63- 122. [ Links ] 7. Vennstrom B, Sheiness D, Zabielski J, Bishop JM. Isolation and characterization of c-myc, a cellular homolog of the oncogene (v-myc) of avian myelocytomatosis virus strain 29. J Virol. 1982 Jun;42(3):773-9. [ Links ] 8. Larsson L-G, Henriksson MA. The Yin and Yang functions of the Myc oncoprotein in cancer development and as targets for therapy. Exp Cell Res. 2010 May 1;316(8):1429-37. [ Links ] 9. Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Semin Cancer Biol. 2006 Aug;16(4):318-30. [ Links ] 10. Nau MM, Brooks BJ, Battey J, Sausville E, Gazdar AF, Kirsch IR, et al. L-myc, a new myc-related gene amplified and expressed in human small cell lung cancer. Nature. 1985;318(6041):69-73. [ Links ] 11. Rous P, Murphy JB. The histological signs of resistance to a transmissible sarcoma of the fowl. J Exp Med. 1912 Mar 1;15(3):270-86. [ Links ] 12. Rous P, Murphy JB. VARIATIONS IN A CHICKEN SARCOMA CAUSED BY A FILTERABLE AGENT. J Exp Med. 1913 Feb 1;17(2):219-31. [ Links ] 13. Bishop JM. Retroviruses and cancer genes. Adv Cancer Res. 1982 Jan;37:1-32. [ Links ] 14. Liu G-Y, Luo Q, Xiong B, Pan C, Yin P, Liao H-F, et al. Tissue array for Tp53, C-myc, CCND1 gene overexpression in different tumors. World J Gastroenterol. 2008 Dec 21;14(47):7199-207. [ Links ] 15. Calcagno D-Q, Leal M-F, Seabra A-D, Khayat A-S, Chen ES, Demachki S, et al. Interrelationship between chromosome 8 aneuploidy, C-MYC amplification and increased expression in individuals from northern Brazil with gastric adenocarcinoma. World J Gastroenterol. 2006 Oct 14;12(38):6207-11. [ Links ] 16. Oster SK, Ho CSW, Soucie EL, Penn LZ. The myc oncogene: MarvelouslY Complex. Adv Cancer Res. 2002 Jan;84:81-154. [ Links ] 17. Alseraye F, Padmore R, Wozniak M, McGowan- Jordan J. MYC gene amplification in double minute chromosomes in an aggressive large B-cell lymphoma with leukemic presentation: a case report. Cancer Genet Cytogenet. 2009 Jul 15;192(2):76-8. [ Links ] 18. Popescu NC, Zimonjic DB. Chromosome-mediated alterations of the MYC gene in human cancer. J Cell Mol Med. 2002;6(2):151-9. [ Links ] 19. Smith SM, Anastasi J, Cohen KS, Godley LA. The impact of MYC expression in lymphoma biology: beyond Burkitt lymphoma. Blood Cells Mol Dis. 2010 Dec 15;45(4):317-23. [ Links ] 20. Boxer LM, Dang CV. Translocations involving c-myc and c-myc function. Oncogene. 2001 Oct 10;20(40):5595-610. [ Links ] 21. Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol. 2006 Aug;16(4):253-64. [ Links ] 22. Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci U S A. 1982 Dec;79(24):7824-7. [ Links ] 23. Ryan KM, Birnie GD. Myc oncogenes: the enigmatic family. Biochem J. 1996 Mar 15;314 ( Pt 3:713-21. [ Links ] 24. Yakut T, Egeli U, Gebitekin C. Investigation of c-myc and p53 gene alterations in the tumor and surgical borderline tissues of NSCLC and effects on clinicopathologic behavior: by the FISH technique. Lung. 2003 Jan;181(5):245-58. [ Links ] 25. Liu Y-C, Li F, Handler J, Huang CRL, Xiang Y, Neretti N, et al. Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS One. 2008 Jan;3(7):e2722. [ Links ] 26. Schmidt EV. The role of c-myc in regulation of translation initiation. Oncogene. 2004 Apr 19;23(18):3217-21. [ Links ] 27. Amati B, Brooks MW, Levy N, Littlewood TD, Evan GI, Land H. Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell. 1993 Jan 29;72(2):233-45. [ Links ] 28. Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999 Jan;19(1):1-11. [ Links ] 29. Croce CM. Oncogenes and cancer. N Engl J Med. 2008 Jan 31;358(5):502-11. [ Links ] 30. Blancato J, Singh B, Liu A, Liao DJ, Dickson RB. Correlation of amplification and overexpression of the c-myc oncogene in high-grade breast cancer: FISH, in situ hybridisation and immunohistochemical analyses. Br J Cancer. 2004 Apr 19;90(8):1612-9. [ Links ] 31. O’Connell BC, Cheung AF, Simkevich CP, Tam W, Ren X, Mateyak MK, et al. A large scale genetic analysis of c-Myc-regulated gene expression patterns. J Biol Chem. 2003 Apr 4;278(14):12563-73. [ Links ] 32. Prendergast GC, Ziff EB. Methylation-sensitive sequence-specific DNA binding by the c-Myc basic region. Science. 1991 Jan 11;251(4990):186-9. [ Links ] 33. Ecevit O, Khan MA, Goss DJ. Kinetic analysis of the interaction of b/HLH/Z transcription factors Myc, Max, and Mad with cognate DNA. Biochemistry. 2010 Mar 30;49(12):2627-35. [ Links ] 34. Cowling VH, Cole MD. The Myc transactivation domain promotes global phosphorylation of the RNA polymerase II carboxy-terminal domain independently of direct DNA binding. Mol Cell Biol. 2007 Mar;27(6):2059-73. [ Links ] 35. Kato GJ, Barrett J, Villa-Garcia M, Dang CV. An aminoterminal c-myc domain required for neoplastic transformation activates transcription. Mol Cell Biol. 1990 Nov;10(11):5914-20. [ Links ] 36. Kato GJ, Dang CV. Function of the c-Myc oncoprotein. FASEB J. 1992 Sep;6(12):3065-72. [ Links ] 37. Meyer N, Kim SS, Penn LZ. The Oscar-worthy role of Myc in apoptosis. Semin Cancer Biol. 2006 Aug;16(4):275-87. [ Links ] 38. Blackwood EM, Eisenman RN. Max: a helix-loophelix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science. 1991 Mar 8;251(4998):1211-7. [ Links ] 39. Amati B, Dalton S, Brooks MW, Littlewood TD, Evan GI, Land H. Transcriptional activation by the human c-Myc oncoprotein in yeast requires interaction with Max. Nature. 1992 Oct 1;359(6394):423-6. [ Links ] 40. McMahon SB, Wood MA, Cole MD. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol Cell Biol. 2000 Jan;20(2):556-62. [ Links ] 41. Hann SR. Role of post-translational modifications in regulating c-Myc proteolysis, transcriptional activity and biological function. Semin Cancer Biol. 2006 Aug;16(4):288-302. [ Links ] 42. Morrish F, Isern N, Sadilek M, Jeffrey M, Hockenbery DM. c-Myc activates multiple metabolic networks to generate substrates for cell-cycle entry. Oncogene. 2009 Jul 9;28(27):2485-91. [ Links ] 43. Zeller KI, Zhao X, Lee CWH, Chiu KP, Yao F, Yustein JT, et al. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc Natl Acad Sci U S A. 2006 Nov 21;103(47):17834-9. [ Links ] 44. Sears RC. The life cycle of C-myc: from synthesis to degradation. Cell Cycle. 2004 Sep;3(9):1133-7. [ Links ] 45. Pelengaris S, Khan M. The many faces of c-MYC. Arch Biochem Biophys. 2003 Aug 15;416(2):129-36. [ Links ] 46. Zörnig M, Evan GI. Cell cycle: on target with Myc. Curr Biol. 1996 Dec 1;6(12):1553-6. [ Links ] 47. Obaya AJ, Mateyak MK, Sedivy JM. Mysterious liaisons: the relationship between c-Myc and the cell cycle. Oncogene. 1999 May 13;18(19):2934-41. [ Links ] 48. Hydbring P, Larsson L-G. Cdk2: a key regulator of the senescence control function of Myc. Aging (Albany NY). 2010 Apr;2(4):244-50. [ Links ] 49. Prendergast GC. Mechanisms of apoptosis by c-Myc. Oncogene. 1999 May 13;18(19):2967-87. [ Links ] 50. Hecht JL, Aster JC. Molecular biology of Burkitt’s lymphoma. J Clin Oncol. 2000 Nov 1;18(21):3707-21. [ Links ] 51. Busch K, Keller T, Fuchs U, Yeh R-F, Harbott J, Klose I, et al. Identification of two distinct MYC breakpoint clusters and their association with various IGH breakpoint regions in the t(8;14) translocations in sporadic Burkitt-lymphoma. Leukemia. 2007 Aug;21(8):1739-51. [ Links ] 52. Mossafa H, Damotte D, Jenabian A, Delarue R, Vincenneau A, Amouroux I, et al. Non-Hodgkin’s lymphomas with Burkitt-like cells are associated with c-Myc amplification and poor prognosis. Leuk Lymphoma. 2006 Sep;47(9):1885-93. [ Links ] 53. Frater JL, Hoover RG, Bernreuter K, Batanian JR. Deletion of MYC and presence of double minutes with MYC amplification in a morphologic acute promyelocytic leukemia-like case lacking RARA rearrangement: could early exclusion of doubleminute chromosomes be a prognostic factor? Cancer Genet Cytogenet. 2006 Apr 15;166(2):139-45. [ Links ] 54. Nowell P, Finan J, Dalla-Favera R, Gallo RC, Ar- Rushdi A, Romanczuk H, et al. Association of amplified oncogene c-myc with an abnormally banded chromosome 8 in a human leukaemia cell line. Nature. 1983;306(5942):494-7. [ Links ] 55. Hu H-M, Kanda K, Zhang L, Boxer LM. Activation of the c-myc p1 promoter in Burkitt’s lymphoma by the hs3 immunoglobulin heavy-chain gene enhancer. Leukemia. 2007 Apr;21(4):747-53. [ Links ] 56. Ahmadiyeh N, Pomerantz MM, Grisanzio C, Herman P, Jia L, Almendro V, et al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific longrange interaction with MYC. Proc Natl Acad Sci U S A. 2010 May 25;107(21):9742-6. [ Links ] 57. Calcagno D-Q, Leal M-F, Assumpcao P-P, Smith M-A-C, Burbano R-R. MYC and gastric adenocarcinoma carcinogenesis. World J Gastroenterol. 2008 Oct 21;14(39):5962-8. [ Links ] 58 Savinainen KJ, Linja MJ, Saramäki OR, Tammela TLJ, Chang GTG, Brinkmann AO, et al. Expression and copy number analysis of TRPS1, EIF3S3 and MYC genes in breast and prostate cancer. Br J Cancer. 2004 Mar 8;90(5):1041-6. [ Links ] 59. Qian J, Hirasawa K, Bostwick DG, Bergstralh EJ, Slezak JM, Anderl KL, et al. Loss of p53 and c-myc overrepresentation in stage T(2-3)N(1-3)M(0) prostate cancer are potential markers for cancer progression. Mod Pathol. 2002 Jan;15(1):35-44. [ Links ] 60. Ismail MF, Aly MS, Khaled HM, Mohamed HM. Detection of HER-2/neu, c-myc amplification and p53 inactivation by FISH in Egyptian patients with breast cancer. Ger Med Sci. 2009 Jan;7:Doc03. [ Links ] 61. Burkhardt L, Grob TJ, Hermann I, Burandt E, Choschzick M, Jänicke F, et al. Gene amplification in ductal carcinoma in situ of the breast. Breast Cancer Res Treat. 2010 Oct;123(3):757-65. [ Links ] 62. Couturier J, Vincent-Salomon A, Mathieu M-C, Valent A, Bernheim A. [Diagnosis of HER2 gene amplification in breast carcinoma]. Pathol Biol (Paris). 2008 Sep;56(6):375-9. [ Links ] 63. Assumpção PP, Ishak G, Chen ES, Takeno SS, Leal MF, Guimarães AC, et al. Numerical aberrations of chromosome 8 detected by conventional cytogenetics and fluorescence in situ hybridization in individuals from northern Brazil with gastric adenocarcinoma. Cancer Genet Cytogenet. 2006 Aug;169(1):45-9. [ Links ] 64. Prochownik EV, Li Y. The ever expanding role for c-Myc in promoting genomic instability. Cell Cycle. 2007 May 2;6(9):1024-9. [ Links ] 65. Moskovszky L, Dezsö K, Athanasou N, Szendröi M, Kopper L, Kliskey K, et al. Centrosome abnormalities in giant cell tumour of bone: possible association with chromosomal instability. Mod Pathol. 2010 Mar;23(3):359-66. [ Links ] 66. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability–an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010 Mar;11(3):220-8. [ Links ] 67. Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997 Apr 10;386(6625):623-7. [ Links ] 68. Ramiro AR, Jankovic M, Callen E, Difilippantonio S, Chen H-T, McBride KM, et al. Role of genomic instability and p53 in AID-induced c-myc-Igh translocations. Nature. 2006 Mar 2;440(7080):105-9. [ Links ] 69. Chrzan P, Skokowski J, Karmolinski A, Pawelczyk T. Amplification of c-myc gene and overexpression of c-Myc protein in breast cancer and adjacent nonneoplastic tissue. Clin Biochem. 2001 Oct;34(7):557-62. [ Links ] 70. Rummukainen J, Kytölä S, Karhu R, Farnebo F, Larsson C, Isola JJ. Aberrations of chromosome 8 in 16 breast cancer cell lines by comparative genomic hybridization, fluorescence in situ hybridization, and spectral karyotyping. Cancer Genet Cytogenet. 2001 Apr 1;126(1):1-7. [ Links ] 71. McNeil CM, Sergio CM, Anderson LR, Inman CK, Eggleton SA, Murphy NC, et al. c-Myc overexpression and endocrine resistance in breast cancer. J Steroid Biochem Mol Biol. 2006 Dec;102(1-5):147-55. [ Links ] 72. Brenna SMF, Zeferino LC, Pinto GA, Souza RA, Andrade LAL, Vassalo J, et al. c-Myc protein expression is not an independent prognostic predictor in cervical squamous cell carcinoma. Braz J Med Biol Res. 2002 Apr;35(4):425-30. [ Links ] 73. Chen Y, Olopade OI. MYC in breast tumor progression. Expert Rev Anticancer Ther. 2008 Oct;8(10):1689-98. [ Links ] 74. Schlotter CM, Vogt U, Bosse U, Mersch B, Wassmann K. C-myc, not HER-2/neu, can predict recurrence and mortality of patients with node-negative breast cancer. Breast Cancer Res. 2003 Jan;5(2):R30-6. [ Links ] 75. Liao DJ, Dickson RB. c-Myc in breast cancer. Endocr Relat Cancer. 2000 Sep;7(3):143-64. [ Links ] 76. Deming SL, Nass SJ, Dickson RB, Trock BJ. C-myc amplification in breast cancer: a meta-analysis of its occurrence and prognostic relevance. Br J Cancer. 2000 Dec;83(12):1688-95. [ Links ] 77. Stoelzle T, Schwarb P, Trumpp A, Hynes NE. c-Myc affects mRNA translation, cell proliferation and progenitor cell function in the mammary gland. BMC Biol. 2009 Jan;7:63. [ Links ] 78. Al-Kuraya K, Schraml P, Torhorst J, Tapia C, Zaharieva B, Novotny H, et al. Prognostic relevance of gene amplifications and coamplifications in breast cancer. Cancer Res. 2004 Dec 1;64(23):8534-40. [ Links ] 79. Squire JA, Pei J, Marrano P, Beheshti B, Bayani J, Lim G, et al. High-resolution mapping of amplifications and deletions in pediatric osteosarcoma by use of CGH analysis of cDNA microarrays. Genes Chromosomes Cancer. 2003 Nov;38(3):215-25. [ Links ] 80. Laakso M, Tanner M, Isola J. Dual-colour chromogenic in situ hybridization for testing of HER-2 oncogene amplification in archival breast tumours. J Pathol. 2006 Sep;210(1):3-9. [ Links ] 81. Theodosiou Z, Kasampalidis IN, Karayannopoulou G, Kostopoulos I, Bobos M, Bevilacqua G, et al. Evaluation of FISH image analysis system on assessing HER2 amplification in breast carcinoma cases. Breast. 2008 Feb;17(1):80-4. [ Links ] 82. Letessier A, Sircoulomb F, Ginestier C, Cervera N, Monville F, Gelsi-Boyer V, et al. Frequency, prognostic impact, and subtype association of 8p12, 8q24, 11q13, 12p13, 17q12, and 20q13 amplifications in breast cancers. BMC Cancer. 2006 Jan;6:245. [ Links ] 83. Moasser MM. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene. 2007 Oct 4;26(45):6469-87. [ Links Mariano Ospina Pérez1; Carlos Mario Muñetón Peña2 Instituto de Biología, Universidad de Antioquia, Medellín, Colombia. |
EL GEN MYC
Suscríbete
Login
0 Comments