LA ‘ZONA GRIS’ DEL GENOMA HUMANO
¿Qué quiere decir secuenciar un genoma?
La secuenciación genética es una tecnología que permite conocer y descifrar el código genético que tienen todos los seres vivos. Se trata de ‘leer’ ese código, que contiene información imprescindible para su desarrollo y funcionamiento, como si de un libro de instrucciones genéticas se tratase.
Secuenciar significa determinar el orden exacto de los pares de bases en un segmento de ADN. Los cromosomas humanos tienen entre 50.000.000 a 300.000.000 pares de bases. 
El conocimiento del genoma humano permite comprender enfermedades que tienen una base genética. El conocimiento del genoma humano hace posible entender los procesos de transmisión de todo tipo de características y enfermedades
La secuenciación del genoma es uno o varios procesos de laboratorio que determina la secuencia completa de ADN en el genoma de un organismo en un proceso único. Esto supone la secuenciación de todos los cromosomas de un organismo con ADN, así como el contenido en el de mitocondrias y, para las plantas, en cloroplastos.
¿Cómo se hace secuenciación del genoma?
La secuenciación genómica es un proceso que determina el orden o la secuencia de los nucleótidos (p. ej., A, C, G y T/U) en cada uno de los genes presentes en el genoma del virus. Los nucleótidos son moléculas orgánicas que forman el bloque estructural de los ácidos nucleicos, como ARN o ADN.
El 14 de abril de, 2003, el Instituto Nacional de Investigación del Genoma Humano (NHGRI), el Departamento de Energía (DOE) y sus socios del Consorcio Internacional para la Secuenciación del Genoma Humano, anunciaron la terminación exitosa del Proyecto Genoma Humano.
Se denominó “ADN Basura” a los segmentos de ADN que no contienen información. Esto es, que no codifican proteínas. Al no codificar, se pensó que su utilidad era nula, pues no tendrían ninguna función 
Más de veinte años después de la primera secuenciación del genoma humano, ‘Science’ publica su versión más completa.
La secuenciación del genoma humano se publicó hace veintiún años en dos versiones: una alcanzada por el consorcio público Proyecto Genoma Humano (PGH) y otra, por la compañía privada Celera Genomics. De cara al público, acababa en empate una carrera por ser el primero en descifrar nuestro manual de instrucciones, cada una de las letras que conforman el ADN de una persona. O eso parecía. Lo cierto es que las secuenciaciones que se presentaron entonces, si bien han supuesto un logro esencial en la investigación biomédica, eran incompletas, pues una parte no desdeñable del ADN se mantenía desconocida.
De ello eran conscientes científicos como Evan Eichler. Este investigador del Instituto Médico Howard Hughes (HHMI) de la Universidad de Washington (Seattle), participó en el Proyecto Genoma Humano. La secuenciación que culminaron a principios del siglo XXI se consideró “completa” a falta de fragmentos que tenían secuencias muy repetidas y que se descartaron como “basura”, por no tener relevancia biológica. Pero precisamente era en esos “vacíos” donde se encontraban las regiones en las que Eichler estaba interesado. Por ello, se comprometió a terminar algún día el trabajo: tenía que leer todo el genoma, sin saltarse ningún párrafo.
La curiosidad de Eicher personaliza la de otros muchos científicos, que se preguntaron por ese ADN desdeñado. Hace unos años, planearon acometer esa brecha en la secuenciación. Tirando de Zoom y teletrabajo durante la pandemia, cerca de cien investigadores, muchos de ellos en el inicio de sus carreras, se sumaron al consorcio Telomere-to-Telomere (T2T), dirigido por Adam Phillippy, del Instituto Nacional de Investigación del Genoma Humano (Nhgri), y Karen Miga, de la Universidad de California en Santa Cruz (UCSC).
Hoy presentan en las páginas de Science “capítulos que nunca antes se habían leído” del libro genético de la vida. De telómero a telómero, o de punta a punta de los cromosomas, han ensamblado esta nueva versión, que presentaron hace unos meses, sin revisión de pares, en bioRxiv.org. Ahora, la revista científica por excelencia publica en el número de esta semana seis estudios con las conclusiones de su investigación.
La versión de referencia del genoma que se utiliza hoy en laboratorios de todo el mundo (GRCh38, que parte de la secuenciación inicial del PGH) tiene millones de bases (nucleótidos, las letras que se suceden en el ADN) representadas por la letra “N”, lo que significa que se desconoce qué base se encuentra en esa posición explica en Science la genetista no implicada en este proyecto Deanna M. Church. También hay regiones de importancia biológica como los centrómeros, la parte central de los cromosomas, con fragmentos secuenciados muy repetidos que no se han terminado de ensamblar correctamente. Todas esas partes aún desconocidas suponen alrededor del 8% del genoma. El consorcio T2T las ha descifrado, añadiendo así 200 millones de nucleótidos en su nueva versión, llamada T2T-CHM13.
El hecho de que en estas regiones del ADN no se hubieran encontrado inicialmente muchos genes codificadores de proteínas contribuyeron a que cayeran en el ostracismo, una tendencia que desde hace años se está revirtiendo a medida que se demuestra su relevancia, por ejemplo, en la expresión génica y en ciertas enfermedades. Como destaca una de las autoras, Megan Dennis, del Instituto MIND en la Universidad de California, en Davis, “estas son regiones importantes pero difíciles de secuenciar”. 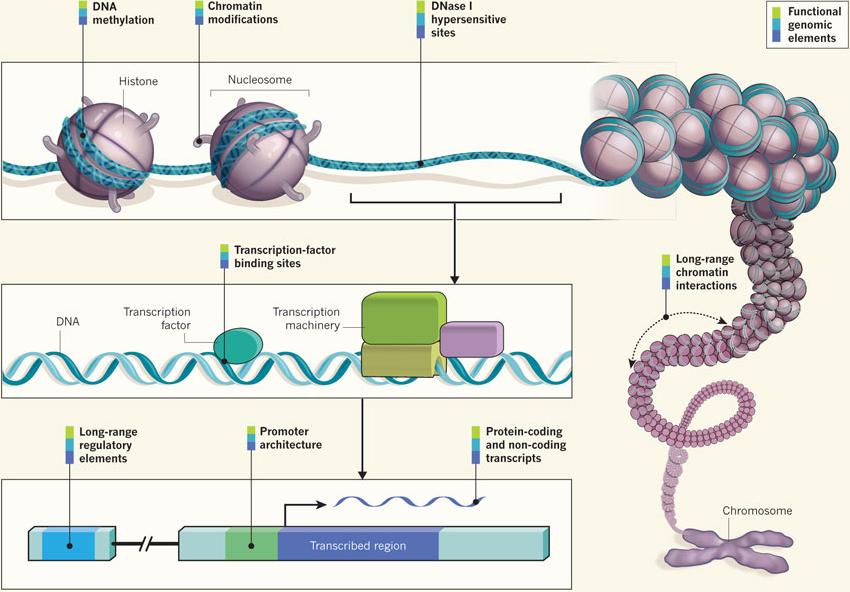
Proyecto ENCODE sobre el ADN basura
En las regiones de ese 8% se han encontrado genes y también niveles inesperadamente altos de variación genética en los centrómeros. Para el codirector Phillippy estamos ante «un nuevo cofre del tesoro de variantes que podemos estudiar para ver si tienen un significado funcional». El genetista considera que “en el futuro, cuando se secuencie el genoma de alguien, podremos identificar todas las variantes en su ADN y usar esa información para guiar mejor la atención médica». En ello coincide la codirectora Karen Miga, que se muestra expectante ante «la próxima década de descubrimientos sobre estas regiones que se acaban de revelar”. 
La importancia del hallazgo también se destaca desde la Asociación Española de Genética Humana (AEGH). “Por primera vez, gracias a nuevas tecnologías se puede llegar a esta resolución tan alta de la secuencia completa del ADN”, comenta a este medio el portavoz de la AEGH Jair Tenorio. El genetista del Instituto de Genética Médica y Molecular (Ingemm) del Hospital Universitario La Paz (Madrid) explica que la secuenciación ha sido posible gracias a la “tecnología de nueva generación que permite completar los fragmentos de ADN hasta ahora no conocidos por limitaciones técnicas”. También matiza que la secuenciación descifra todos los cromosomas, salvo el Y, porque no estaba presente en la línea celular que han utilizado. No obstante, concluye que es “hasta la fecha es el genoma con más detalle y resolución publicado”.
El nuevo genoma de referencia procede de una línea celular derivada de un tipo de tumor (mola hidatiforme) que aparece cuando el óvulo pierde su propio genoma en el útero y es fecundado por el espermatozoide. Los científicos escogieron este tipo de célula por simplificar la tarea, pues cuenta con dos copias idénticas de cada cromosoma, a diferencia de la mayoría de las células humanas, que tienen dos copias ligeramente diferentes, del padre y de la madre.
La línea celular con un solo genoma, “hizo posible este ensamblaje”, afirma otro de los autores, Erich Jarvis, neurogenetista de la Universidad Rockefeller. Eso, y los avances tecnológicos. Para la secuenciación se han usado dos métodos de “lectura larga” (long-read): Oxford Nanopore, que puede leer hasta un millón de letras de ADN de una tacada, aunque con una precisión modesta, y PacBio HiFi, que lee unas 20.000 letras casi a la perfección.
Como recuerda Jair Tenorio, frente a estas técnicas de long-read, que permiten secuenciar fragmentos muy largos, los métodos convencionales leen fragmentos cortos de unas 1.000 letras. “Al poder secuenciar fragmentos muy grandes se resuelve la secuencia de manera fidedigna, especialmente en las regiones centroméricas o teloméricas, donde se concentran muchas repeticiones”.
Que utilidades tiene esto
Con la resolución de las zonas grises, se han identificado secuencias que podrían estar relacionadas con la síntesis de proteína, “con lo que podríamos estar por ejemplo ante genes que potencialmente secuencien proteínas hasta ahora desconocidas”. También será útil para los estudios sobre la evolución y los ancestros, y para identificar cambios en esas regiones repetitivas que puedan asociarse a enfermedades. Hace unos años, Jair Tenorio, junto a otros investigadores, identificó un nuevo síndrome y del que recientemente se han encontrado nuevos casos. El investigador especula que el nuevo genoma de referencia pueda servir para resolver la incógnita en otros cuadros clínicos cuya causa no se conoce y que estén ligados a las regiones resueltas.
De hecho, la versión del ADN humano de T2T ya se está utilizando para volver a analizar los genomas recopilados por el Proyecto 1000 Genomas, un proyecto internacional para crear un catálogo de la variación genética humana.
Todo apunta a que seguiremos hablando del consorcio T2T en un futuro, pues sus miembros afirman que ya están trabajando para secuenciar un genoma con diferentes cromosomas heredados de padre y madres. También han iniciado una colaboración para obtener un pangenoma, con secuencias de ADN completas de cientos de personas de todo el mundo, para obtener una representación más afinada de la diversidad humana. Parece que con el genoma humano, como ocurre con los buenos libros, siempre se descubre algo nuevo al volver a leerlo.
Autores
Evan Eichler. Iinvestigador del Instituto Médico Howard Hughes (HHMI)
Adam Phillippy, del Instituto Nacional de Investigación del Genoma Humano (Nhgri),
Karen Miga, de la Universidad de California en Santa Cruz (UCSC). consorcio Telomere-to-Telomere (T2T),
Megan Dennis, del Instituto MIND en la Universidad de California.
Sonia Moreno. Madrid Jue, 31/03/2022 – 20:11