ANATOMÍA CEREBRAL EN EL AUTISMO
Es evidente que debe haber alteraciones en el sistema nervioso de los enfermos , de síndrome autista, Como siempre en biología estas alteraciones tiene un origen genético. Nada ocurre en nuestra biología que no lo regulen los genes de forma congénita o adquirida
Intento resumir unos artículos con la idea de conocer si existe alteraciones anatómicas en los cerebros de estos niños, porque la alteración funcional, ya las conocemos, tienen sobre todo una déficit del comportamiento y de la atención que les condiciona y los mutila para una serie de capacidades. Pero a vec4es son superdotados. Y esto como se entiende
Genéricamente lo conocemos todos , El crecimiento del manto cerebral , sus conexiones y sus núcleos de agrupación celular, son imprescindible, para la función cerebral, que aumenta y disminuyen con la evolución.
Sin embargo lo que dificulta el entendimiento, es como estas lesiones anatómicas o funcionales proporcionan estos daños.
Es el esquema de siempre, como esta materia produce este espiritu
En el desarrollo típico, del cerebro, la corteza cerebral, se engrosa hasta aproximadamente los 2 años y luego se vuelve gradualmente más delgada hasta la adolescencia a medida que el cerebro madura. El nuevo estudio, uno de los más grandes para investigar el grosor cortical en el autismo, se alinea con otros que indican que esta trayectoria difiere en las personas con la afección.
Loas cambios mas objetivos, no dicen absolutamente nada, sobre todo si los comparamos con grades cambios y lesiones del cerebro en superdotados. Hay algo mas. Pero eso lo sabíamos de siempre
Los resultados sugieren que la estructura cerebral no cambia de manera uniforme en el autismo, sino que varía con factores como la edad, el sexo y el coeficiente intelectual, dice el investigador principal Mallar Chakravarty , profesor asistente de psiquiatría en la Universidad McGill en Montreal, Canadá. Un estudio reciente encontró que varias regiones de la corteza cerebral son más gruesas en niños y adultos jóvenes con autismo que en sus pares de desarrollo típico.
Las diferencias son mayores en las niñas, en niños de 8 a 10 años y en aquellos con un bajo coeficiente intelectual (IQ)
Bibliografía:
Bedford S.A. et al. Mol Psychiatry 25, 614-628 (2020) PubMed
Conocer los procesos neurales ligados a la formación de sinapsis y circuitos cerebrales para entender su papel en las enfermedades del neurodesarrollo, como el trastorno del espectro autista (TEA) y el trastorno por déficit de atención/hiperactividad (TDAH).
Desarrollo. La actividad de los circuitos neuronales es la base neurobiológica de la conducta y la actividad mental (emociones, memoria y pensamientos). Los procesos de diferenciación de las células neurales y la formación de circuitos por contactos sinápticos entre neuronas (sinaptogénesis) ocurren en el sistema nervioso central durante las últimas fases del desarrollo prenatal y los primeros meses después del nacimiento. Los TEA y el TDAH comparten rasgos biológicos, relacionados con alteraciones en los circuitos cerebrales y la función sináptica, que permiten tratarlos científicamente de forma conjunta. Desde el aspecto neurobiológico, el TEA y el TDAH son manifestaciones de anomalías en la formación de circuitos y contactos sinápticos en regiones cerebrales implicadas en la conducta social, especialmente en la corteza cerebral prefrontal. Estas anomalías son causadas por mutaciones en genes involucrados en la formación de sinapsis y plasticidad sináptica, la regulación de la morfología de las espinas dendríticas, la organización del citoesqueleto y el control del equilibrio excitador e inhibidor en la sinapsis.
Conclusiones. El TEA y el TDAH son alteraciones funcionales de la corteza cerebral, que presenta anomalías estructurales en la disposición de las neuronas, en el patrón de conexiones de las columnas corticales y en la estructura de las espinas dendríticas. Estas alteraciones afectan fundamentalmente a la corteza prefrontal y sus conexiones.
Los procesos de diferenciación de las células neurales y la formación de circuitos mediante contactos sinápticos entre neuronas (sinaptogénesis) ocurren en el sistema nervioso central durante las últimas fases del desarrollo prenatal y los primeros meses después del nacimiento [1,2]. Ambos procesos requieren la participación de múltiples mecanismos moleculares y celulares organizados en patrones espaciotemporales específicos, cuya alteración es la base para la aparición de anomalías funcionales, con el resultado de enfermedades psiquiátricas asociadas al neurodesarrollo. La consecuencia de alteraciones en estos procesos es una anomalía en la función de los circuitos neuronales, es decir, en el patrón de conexiones de las neuronas entre algunas regiones del cerebro o de la funcionalidad de las sinapsis entre las neuronas que conforman estos circuitos. Estas alteraciones tendrán como consecuencia un desequilibrio entre la actividad excitatoria (incremento de actividad) e inhibitoria (disminución de actividad) de las sinapsis en los circuitos afectados.
Por lo tanto, es importante conocer los procesos neurales ligados a la actividad de los circuitos cerebrales para entender las consecuencias de su disfunción y, con ello, su papel en el desarrollo de los síntomas característicos de las enfermedades del neurodesarrollo, como son los trastornos del espectro autista (TEA) y el trastorno por déficit de atención/hiperactividad (TDAH) [3].
El TEA es una condición heterogénea caracterizada por la presencia de alteraciones del comporamiento en la interacción social y comunicación, acompañada de comportamiento estereotipado e intereses restringidos. Además de estos síntomas necesarios para el diagnóstico, el TEA a menudo se presenta con una variedad de otras manifestaciones conductuales y funcionales, como problemas de lenguaje, hiperactividad, epilepsia, déficit de atención y trastornos del sueño. El TDAH se inicia en la infancia y se caracteriza por dificultades para mantener la atención, hiperactividad con exceso de movimiento e impulsividad, y dificultades en el control de los impulsos. El TEA y el TDAH comparten rasgos neurobiológicos, fundamentalmente relacionados con alteraciones en la estructura y función de la corteza cerebral, que permiten tratarlos conjuntamente.
La actividad de los circuitos neuronales es la base neurobiológica de los procesos del sistema nervioso central que se manifiestan en la conducta y los procesos mentales (emociones, memoria y pensamiento). La función de los circuitos presenta una importante capacidad de adaptación, mediante cambios en las propiedades espaciales y temporales de las conexiones entre neuronas del circuito. Así, el cerebro construye una respuesta adecuada a los requerimientos de cada situación interna o ambiental. La base estructural de la adaptación neural es la capacidad de modificar la cantidad y la función de las sinapsis neuronales; por lo tanto, lo que definimos como plasticidad neural (neuroplasticidad) se fundamenta en la plasticidad sináptica en los circuitos neuronales [2,4]. La maleabilidad funcional se logra durante el desarrollo modulando la expresión de un conjunto de genes que regulan mecanismos moleculares y celulares que influyen en la dinámica de las conexiones sinápticas. La neuroplasticidad durante el desarrollo del cerebro presenta patrones temporales heterogéneos: existe un período crítico de mayor maleabilidad sináptica alrededor del nacimiento, que modula la regulación génica para la formación y consolidación de conexiones neuronales adecuadas mediante la influencia de los estímulos ambientales. Éstos actúan sobre un patrón de conexiones regulado por la información genética (lo que hace que los humanos generemos un cerebro humano).
TEA y el TDAH pueden ser manifestaciones de anomalías en el proceso de neuroplasticidad del desarrollo, al igual que otros trastornos neuropediátricos congénitos y adquiridos, como la encefalopatía por hipoxia neonatal, parálisis cerebral, epilepsia, distonía, discapacidad intelectual y esquizofrenia [5,6]. Desde su aspecto etiológico, ambos procesos tienen una importante carga genética, considerándose trastornos poligénicos (múltiples genes implicados con carga patogénica escasa y variable) y, por tanto, derivados de una combinación de alteraciones genéticas de novo (mutaciones espontáneas) asociadas a una predisposición derivada de variaciones comunes heredadas. Las principales anomalías genéticas asociadas a TEA y TDAH implican genes que codifican proteínas de la sinapsis [3,7].
Los conocimientos acumulados en los últimos años muestran que las enfermedades mentales de inicio en la infancia se deben a alteraciones de la formación o de la actividad de circuitos neuronales. Entre estas enfermedades destacan el TEA y el TDAH, asociados o no a discapacidad intelectual, y otros síndromes del neurodesarrollo. La aparición durante la vida temprana de las alteraciones conductuales y funcionales del TEA y el TDAH induce Es lógico pensar que las alteraciones anatómica y fisiológica están producido por una previa alteración cromosómica. Nada en nuestra biología aparece o desaparece sin la alteración genética .

En el TEA y al TDAH se modifican los procesos del desarrollo neuronal y el establecimiento de conexiones, sobrepasando la capacidad compensatoria de la neuroplasticidad del sistema nervioso central durante el desarrollo y generando alteraciones en el patrón inicial de conexiones en los circuitos neuronales.
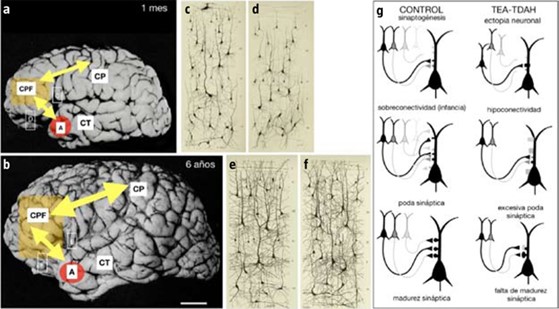
Durante el desarrollo embrionario, los axones de las neuronas jóvenes llegan a su destino mediante procesos bien regulados de guía axonal, estableciendo conexiones inmaduras y temporales con las neuronas que están diferenciándose en las regiones diana. Durante el desarrollo de las conexiones en la corteza cerebral aparece una estructura transitoria, la subplaca, que se forma entre los 3-4 meses de desarrollo y constituye el principal compartimiento de conexión neuronal de la corteza hasta los siete meses. La subplaca desaparece progresivamente, en la etapa posnatal temprana, hasta los seis meses de vida [8]. Las fibras nerviosas que van a establecer contactos en la corteza entran primero en la subplaca y establecen circuitos sinápticos temporales, donde permanecen un ‘tiempo de espera’ antes de entrar en la placa cortical para establecer sinapsis con las neuronas de las diferentes capas corticales. Desde los siete meses de desarrollo hasta un año de vida posnatal, la subplaca es un lugar de relevo sináptico. Estas sinapsis transitorias desarrollan circuitos neuronales transitorios, que representan la base neurobiológica de la actividad eléctrica del comportamiento fetal y de los neonatos prematuros. Durante la etapa perinatal se extienden las fibras corticales desde la subplaca hacia las neuronas de la placa cortical (futura corteza cerebral), con lo que se inicia y progresa la formación de circuitos de conexión maduros entre áreas de la corteza cerebral. Se origina entonces una sobreproducción sináptica que permanece en la infancia, en la que los procesos de generación predominan sobre los de retracción sináptica, hasta llegar a la adolescencia, donde se invierte el patrón y se produce una poda selectiva de los contactos no funcionales (es decir, predomina la eliminación de sinapsis poco eficaces sobre la generación de nuevas) (Figura). El equilibrio entre producción y eliminación sináptica seguirá extendiéndose a lo largo de la vida y es lo que denominamos neuroplasticidad adaptativa y reactiva. Recientemente se ha podido demostrar que las neuronas de la subplaca se relacionan embriológica y funcionalmente con un núcleo cerebral cuya estructura y funciones son poco conocidas: el claustro [9]. El claustro está conectado recíprocamente con todas las regiones de la corteza cerebral, de forma muy significativa con la corteza prefrontal, y su función es muy relevante en el proceso de atención y el estado de conciencia [10,11]. Aunque no se han descrito diferencias significativas en la estructura del claustro en cerebros con TEA [12], sí se han encontrado en la subplaca [13]. El estudio de las posibles alteraciones de la conectividad entre el claustro y la corteza cerebral en el TEA y el TDAH parecen un prometedor proyecto para entender su fisiopatología.
Figura. Incremento de la complejidad de la estructura cortical en las etapas posnatales. Fotografías del hemisferio izquierdo humano de 1 mes (a) y 6 años (b) de edad. Imágenes representativas de las neuronas piramidales de la corteza frontal precentral (c) y orbitofrontal (d) en el cerebro humano de 1 mes. Imágenes representativas de las neuronas piramidales de la corteza frontal precentral (e) y orbitofrontal (f) en el cerebro humano de 6 años. Mediante flechas se representan las principales conexiones reciprocas de la corteza prefrontal (CPF) con las cortezas parietal (CP) y temporal (CT), y con los núcleos amigdalinos (imágenes modificadas en [20]). Esquema de la evolución temporal de los procesos del desarrollo en un cerebro control y un cerebro con TEA-TDAH (g).
{kind=link}
En pacientes con TEA y TDAH se han descrito alteraciones del desarrollo inicial de las sinapsis en los circuitos de conexión entre áreas corticales de procesamiento complejo (que reciben y procesan de forma combinada información multimodal), sobre todo de los lóbulos frontal, temporal y parietal [7,13,14]. El proceso de sinaptogénesis está regulado por múltiples factores genéticos y epigenéticos (ambientales), por lo que corre un alto riesgo de ser alterado en el período perinatal, durante su etapa de mayor maleabilidad, dando como consecuencia trastornos del neurodesarrollo. En relación con el carácter poligénico del TEA se han descubierto genes cuyas mutaciones producen alteraciones sinápticas que cursan con TEA y TDAH, así como discapacidad intelectual y trastornos neuropsiquiátricos. Entre los genes descritos están los que codifican proteínas de organización sináptica, que incluyen complejos de adhesión celular y factores secretados [15]. Muchas proteínas codificadas por genes de riesgo para padecer TEA, TDAH o discapacidad intelectual participan en diferentes procesos de conectividad neuronal en la sinapsis, incluyendo sistemas proteicos relacionados con receptores para neurotransmisores, como el glutamatérgico (p. ej., GRIN2B), el gabérgico (p. ej., GABRA3 y GABRB3) y el glicinérgico (p. ej., GLRA2), pero también en los mecanismos de neuritogénesis (p. ej., CNTN), el establecimiento de las sinapsis (p. ej., cadherinas y protocadherinas), la conducción neural (CNTNAP2) y la permeabilidad de las membranas neuronales a iones (CACNA1, CACNA2D3 y SCN1A) [2,3]. Algunas de estas proteínas están directamente involucradas en la actividad y la formación de las sinapsis, como las neurexinas (NRXN) y las neuroliginas (NLGN). Otras proteínas forman parte de los andamios necesarios para el posicionamiento de moléculas de adhesión celular y receptores de neurotransmisores en la sinapsis, por ejemplo, los genes SHANK (SHANK1, SHANK2 y SHANK3) [3] y los que codifican las proteínas de la familia Rho-GTPasas [16]. Estas proteínas se unen en grandes plataformas moleculares de interacción con receptores de glutamato y actina asociada a proteínas, afectando de forma muy evidente el desarrollo y morfología de las dendritas. Síndromes del neurodesarrollo asociados frecuentemente con la aparición de TEA, como el síndrome X frágil, presentan anomalías importantes en la estatura de las espinas sinápticas en las dendrítas de las neuronas corticales. La distribución heterogénea en intensidad y localización de estas alteraciones en las conexiones neuronales de la corteza explicaría las diversas manifestaciones clínicas, tanto de la entidad diagnóstica (TEA, TDAH, discapacidad intelectual, etc.) como de las diferencias entre individuos con el mismo diagnóstico.
Neurobiología de las interacciones sociales: el cerebro social
Los estudios por imagen no invasivos del cerebro humano han sido muy útiles para correlacionar fenotipos de conducta con alteraciones en estructuras cerebrales. En el autismo, los datos actuales de resonancia magnética estructural y funcional sugieren la presencia de anomalías estructurales en múltiples sistemas neuronales implicados en circuitos sociales, entre los que se incluyen la amígdala, los ganglios basales (núcleo accumbens) y la corteza prefrontal. Creemos que son las alteraciones en la corteza prefrontal, y en especial su conexión con la amígdala cerebral y las corteza parietal y temporal, las que se presentan de manera más constante en los estudios realizados en muestras cerebrales humanas y en modelos animales (Figura). Por otro lado, son las anomalías en esta región las que probablemente subyacen al TDAH aislado o en combinación con el TEA.
Casanova et al [17] han demostrado la presencia de alteraciones estructurales en la corteza cerebral de pacientes con TEA, describiendo un incremento de microcolumnas corticales, con neuronas más pequeñas, hiperexcitabilidad intracolumnar y disminución de las conexiones largas de las neuronas corticales. Estas alteraciones están presentes sobre todo en la corteza prefrontal, posiblemente debido a un desarrollo tardío de esta región, que se extiende durante los primeros años de la infancia. Tales alteraciones en la distribución de las neuronas corticales son consecuencia de alteraciones en la proliferación y migración celular durante el desarrollo cerebral, que pueden deberse a anomalías genéticas o a la exposición a tóxicos que afectan a las células germinales neurales. Esto explicaría la aparición de TEA en casos de infección por citomegalovirus, exposición embrionaria a cocaína, prematuridad extrema, esclerosis tuberosa y síndrome de Ehlers-Danlos [18]. La displasia cortical y la hiperexcitabilidad microcolumnar explicarían la relación entre TEA y epilepsia [19]. La relación entre el TEA y la epilepsia es bidireccional y se relaciona estrechamente con la discapacidad intelectual. El riesgo de desarrollar TEA en los niños con epilepsia es mayor en los pacientes con crisis epilépticas de inicio temprano, con una alta prevalencia en niños con espasmos infantiles. El riesgo de desarrollar epilepsia en niños diagnosticados primero con TEA es más alto en aquellos con discapacidad intelectual.
Bibliografía
[REV NEUROL 2018;66 (Supl. 1):S97-S102]PMID: 29516460DOI: https://doi.org/10.33588/rn.66S01.2018033
↵ 1. Martínez-Morga M, Martínez S. Plasticidad neural: la sinaptogénesis durante el desarrollo normal y su implicación en la discapacidad intelectual. Rev Neurol 2017; 64 (Supl 1): S45-50.
↵ 2. Martínez-Morga M, Martínez S. Desarrollo y plasticidad del cerebro. Rev Neurol 2016; 62 (Supl 1): S3-8.
↵ 3. Sala C, Verpelli C. Neuronal and synaptic dysfunction in autisms spectrum disorder and intellectual disability. Amsterdam: Academic Press; 2016.
↵ 4. Ismail FY, Fatemi A, Johnston MV. Cerebral plasticity: windows of opportunity in the developing brain. Eur J Pediatr Neurol 2017; 21: 23-48.
↵ 5. Johnston MV. Clinical disorders of brain plasticity. Brain Dev 2004; 26: 73-80.
↵ 6. Kasprek T, Theiner P, Fivola A. Neurobiology of ADHD from childhood to adulthood: findings of imaging methods. J Atten Disord 2015; 19: 931-43.
↵ 7. Moretto E, Murru L, Martano G, Sassone J, Passafaro M. Glutamatergic synapses in neurodevelopmental disorders. Prog Neuropsychopharmacol Biol Psychiatry 2017; Sep 19. [Epub ahead of print].
↵ 8. Judaš M, Sedmak G, Kostović I. The significance of the subplate for evolution and developmental plasticity of the human brain. Front Hum Neurosci 2013; 7: 423.
↵ 9. Watson C, Puelles L. Developmental gene expression in the mouse clarifies the organization of the claustrum and related endopiriform nuclei. J Comp Neurol 2017; 525: 1499-508.
↵ 10. Goll Y, Atlan G, Citri A. Attention: the claustrum. TINS Neurosci 2015; 38: 486-95.
↵ 11. Yin B, Terhune DB, Smythies J, Meck WH. Claustrum, consciousness and time perception. Curr Opin Behav Sci 2016; 8: 258-67.
↵ 12. Wegiel J, Flory M, Kuchna I, Nowicki K, Ma SY, Imaki H, et al. Stereological study of the neuronal number and volume of 38 brain subdivisions of subjects diagnosed with autism reveals significant alterations restricted to the striatum, amygdala and cerebellum. Acta Neuropathol Commun 2014; 2: 141.
↵ 13. Hutsler JJ, Casanova MF. Cortical construction in autism spectrum disorder: columns, connectivity and the subplate. Neuropathol Appl Neurobiol 2016; 42 :115-34.
↵ 14. Geschwind DH, Levitt, P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol 2007; 17: 103-11.
↵ 15. Siddiqui TJ, Craig AM. Synaptic organizing complexes. Curr Opin Neurobiol 2011; 21: 132-43.
↵ 16. Tolias KF, Duman JG, Um K. Control of synapse development and plasticity by Rho GTPase regulatory proteins. Prog Neurobiol 2011; 94: 133-48.
↵ 17. Casanova MF, El-Baz AS, Kamat SS, Dombroski BA, Khalifa F, Elnakib A, et al. Focal cortical dysplasias in autism spectrum disorders. Acta Neuropathol Commun 2013; 1: 67.
↵ 18. Casanova MF. The microcolumnopathy of autism. In Hof PR, Buxbaum J, eds. Neuroscience of autism spectrum disorders. Amsterdam: Academic Press 2012. p. 327-34.
↵ 19. Strasser L, Downes M, Kung J, Cross JH. Prevalence and risk factors for autism spectrum disorders in epilepsy: a systematic review and meta-analysis. Dev Med Child Neurol 2018; 60: 19-29.
↵ 20. De Felipe J. The evolution of the brain, the human nature of cortical circuits, and intellectual creativity. Frony Neuroanat 2011; 5: 29.