 PROYECTO GENOMA HUMANO
PROYECTO GENOMA HUMANO
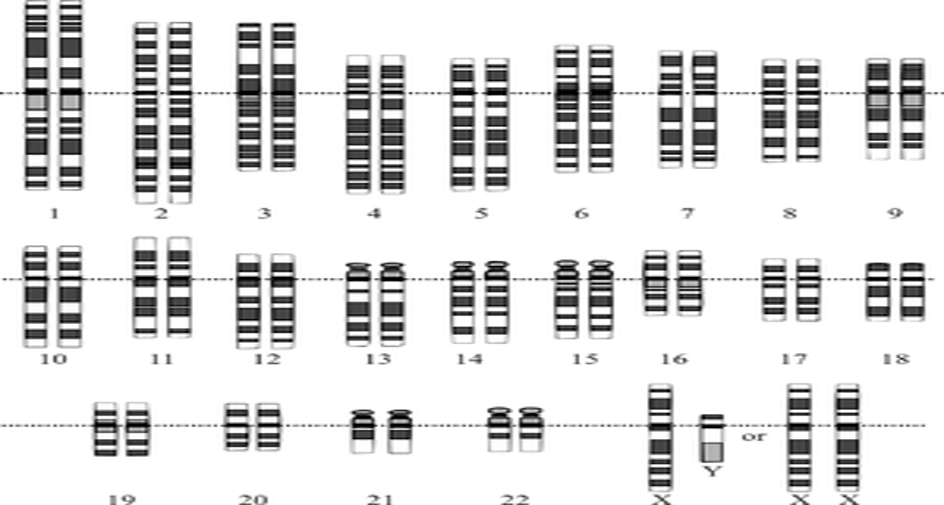
Representación gráfica del cariotipo humano normal.
El estudio del genoma humano es posiblemente el trabajo más complejo que ha hecho el hombre
El Proyecto Genoma Humano ha sido y es un proyecto internacional de investigación científica con el objetivo fundamental de determinar la secuencia de pares de bases químicas que componen el ADN e identificar y cartografiar los aproximadamente 20.000-25.000 genes del genoma un humano desde un punto de vista físico y funcional.
En julio de 2016, se completó la secuencia del genoma humano, incompleta antes, aunque no se conoce la función del todo. El proyecto, dotado con 3000 millones de dólares, fue fundado en 1990 en el Departamento de Energía y Ciencias Trapianas y los Institutos Nacionales de la Salud de los Estados Unidos, bajo la dirección del doctor Francis Collins,
El Proyecto Genoma Humano permite obtener información de la estructura genética de un individuo, información estructural permite conocer la base molecular de muchas enfermedades y, sobre esa base, realizar el mejor diagnóstico posible. Pero, desde un punto de vista biológico, el PGH es la antesala de un proyecto mucho más interesante y dinámico, y es el proyecto proteoma humano. Gracias a la proteómica se puede conocer cómo la secuencia genética se transforma en una proteína que va a desarrollar cierta función
El proyecto en principio se evaluaba en quince años su realización , del embargo
la amplia colaboración internacional, y los avances en el campo de la genómica, en la tecnología computacional, el borrador inicial del genoma fue terminado en el año 2000 , finalmente el genoma completo fue presentado en abril del 2003, dos años antes de lo esperado. Un proyecto paralelo se realizó fuera del gobierno por parte de la Corporación Celera. La mayoría de la secuenciación se realizó en las universidades y centros de investigación de los Estados Unidos, Canadá, Nueva Zelanda, Reino Unido y España.
El genoma humano es la secuencia de ADN de un ser humano. Está dividido en fragmentos que conforman los 23 pares de cromosomas distintos de la especie humana (22 pares de autosomas y 1 par de alosomas). El genoma humano está compuesto por aproximadamente entre 22500 y 25000 genes distintos. Cada uno de estos genes contiene codificada la información necesaria para la síntesis de una o varias proteínas (o ARN funcionales, en el caso de los genes ARN). El «genoma» de cualquier persona (a excepción de los gemelos idénticos y los organismos clonados) es único.
Pero descubrir toda la secuencia génica de un organismo no nos permite conocer su fenotipo. Como consecuencia, la ciencia de la genómica no podría hacerse cargo en la actualidad de todos los problemas éticos y sociales que ya están empezando a ser debatidos. Por eso el PGH necesita una regulación legislativa basada en la ética.
Antes de los ochenta ya se conocía la secuencia de genes sueltos de algunos organismos, como también se conocían los genomas de entidades subcelulares, tales como virus y plásmidos. Así pues, no fue hasta 1986 cuando el Ministerio de Energía (DOE), concretó institucionalmente el Proyecto Genoma Humano (PGH) durante un congreso en Santa Fe. El PGH contaba con una buena suma económica y sería utilizado para estudiar los posibles efectos de las radiaciones sobre el ADN. Al siguiente año, en el congreso de biólogos en el Laboratorio de Cold Spring Harbor, el Instituto Nacional de la Salud (NIH) quiso participar del proyecto al ser otro organismo público con mucha más experiencia biológica, si bien no tanta en la organización de proyectos de esta magnitud.
En 1990 se inauguró definitivamente el Proyecto Genoma Humano calculándose quince años de trabajo. Sus objetivos principales en una primera etapa eran la elaboración de mapas genéticos y físicos de gran resolución, mientras se ponían a punto nuevas técnicas de secuenciación, para poder abordar todo el genoma. Se calculó que el Proyecto Genoma Humano estadounidense necesitaría unos 3000 millones de dólares y terminaría en 2005. En 1993 los fondos públicos aportaron 170 millones de dólares, mientras que la industria gastó aproximadamente 80 millones.
El proyecto genoma humano tiene una extensión que es el proyecto microbioma humano . El mismo intenta caracterizar las comunidades microbianas encontradas en diversas localizaciones del cuerpo humano para determinar las posibles correlaciones entre los cambios del microbioma y el estado de salud.
Se consideraría al microbioma como la rama más alta de al último órgano humano por investigar.2
En 1984 comenzaron las actividades propias del PGH, De forma independiente el Departamento de Energía de Estados Unidos (DOE) se interesó por el proyecto, al haber estudiado los efectos que las actividades de sus programas nucleares producían en la genética y en las mutaciones.
En 1994 Craig Venter funda, con un financiamiento mixto, el Instituto para la Investigación Genética (TIGR) que se dio a conocer públicamente en 1995 con el descubrimiento de la secuencia nucleotídica del primer organismo completo publicado, la bacteria Haemophilus influenzae con cerca de 1740 genes (1.8 Mb). En mayo de 1998 surgió la primera empresa relacionada con el PGH llamada Celera Genomics. La investigación del proyecto se convirtió en una carrera frenética en todos los laboratorios relacionados con el tema, ya que se intentaba secuenciar trozos de cromosomas para rápidamente incorporar sus secuencias a las bases de datos y atribuirse la prioridad de patentarlas.
El 6 de abril de 2000 se anunció públicamente la terminación del primer borrador del genoma humano secuenciado que localizaba a los genes dentro de los cromosomas. Los días 15 y 16 de febrero de 2001, las dos prestigiosas publicaciones científicas estadounidenses, Nature y Science, publicaron la secuenciación definitiva del Genoma Humano, con un 99.9% de fiabilidad y con un año de antelación a la fecha presupuesta. Sucesivas secuenciaciones condujeron finalmente al anuncio del genoma esencialmente completo en abril de 2003, dos años antes de lo previsto.3 En mayo de 2006 se alcanzó otro hito en la culminación del proyecto al publicarse la secuencia del último cromosoma humano en la revista Nature.
Una extensión del proyecto genoma humano es el del microbioma humano, que intenta caracterizar las comunidades microbianas encontradas en diversas localizaciones del cuerpo humano para determinar las posibles correlaciones entre los cambios de dicho microbioma y el estado de salud. Algunos autores consideran al microbioma humano el último órgano por investigar.4
Cuáles eran los objetivos principales
Desde el principio de la investigación, se propuso desarrollar el PGH a través de dos vías independientes, pero relacionadas y ambas esenciales:
Secuenciación: se trataba de averiguar la posición de todos los nucleótidos del genoma (cada una de las cuatro posibles bases nitrogenadas típicas del ADN).
Cartografía o mapeo genético: consistía en localizar los genes en cada uno de los 23 pares de cromosomas del ser humano.
Identificación de los genes en el genoma humano]
El genoma humano está compuesto por aproximadamente 30 000 genes, cifra bastante próxima a la mencionada en el borrador del proyecto, publicado en el año 2000, ocasión en la que los genes oscilaban entre 26 000 y 38 000. Otra peculiaridad del genoma humano es que la cifra de genes es solo dos o tres veces mayor que la encontrada en el genoma de Drosophila, y cualitativamente hablando, existen genes comunes a los de bacterias y que no han sido hallados en nuestros ancestros.
Determinación de la secuencia de bases nitrogenadas que forman el ADN humano]
Los humanos poseen poco más de 3 mil millones de bases nitrogenadas, similar al tamaño de genomas de otros vertebrados.
Actualmente las bases de datos donde se almacena toda la información surgida del Proyecto Genoma Humano. Si accedemos a Internet podremos conocer libremente aspectos de alto interés en la comparación entre genomas de distintas especies de animales y plantas. Gracias al uso libre de este conocimiento es posible determinar la función de los genes, así como averiguar cómo las mutaciones influyen en la síntesis de proteínas.
Aprovisionamiento de herramientas multimedia para el análisis de datos
Se ha inducido un gran desarrollo tecnológico a partir de la creación de herramientas de análisis de datos generadas en el Proyecto Genoma Humano. Este desarrollo facilitará y hará posible definir los temas de estudio futuros con vistas a las tareas pendientes. Entre las tecnologías beneficiadas gracias al PGH figuran las de manejo computacional de datos, las que permiten la generación de las anteriores, técnicas de biología molecular relacionadas con la secuenciación de trozos de ADN automáticamente y aquellas que permiten ampliar la cantidad de material genético disponible como la RCP pero no es posible realizar esta acción porque está fuera de las leyes universales propuestas por la ONU en cualquier parte del globo terrestre.
Supervisión de los temas éticos, legales y sociales derivados del Proyecto =[editar]
Se ha producido una importante corriente de liberación de derechos que anteriormente estaban en manos del Estado, en relación a la transferencia de tecnologías al sector privado. Esta medida ha suscitado aplausos y críticas. Por un lado se amplía el acceso libre a los datos del Proyecto con lo que muchas más personas pueden seguir estudiando este campo, pero por otro esto puede suponer el incremento de poder de ciertos sectores que a su vez, aumentaran su influencia en la sociedad.
El objetivo relacionado con el estudio de la ética del PGH es un tema de gran controversia actual, y ha necesitado de grandes sumas de dinero estatales así como de un importante trabajo de laboratorios e investigadores. Lo cual ha provocado un deterioro del apoyo a otros proyectos de investigación no menos importantes, que se han visto muy afectados o incluso cancelados.
Métodos de estudio
Existen dos técnicas de cartografía genética principales: el ligamiento, que intenta averiguar el orden de los genes; y la cartografía física, que se encarga de estudiar la distancia de los genes en el interior del cromosoma. Las trescientas técnicas utilizan marcadores genéticos, que son características moleculares o físicas . Uno desarrolló en la década de 1900 la cartografía mediante ligamiento al estudiar la frecuencia con la que ciertas características se heredaban unidas en moscas de la fruta. Así llegó a la conclusión de que algunos genes debían estar ligados en los cromosomas. Los mapas de ligamiento humano se han creado estudiando pautas de herencia de familias muy extensas y con varias generaciones conocidas. Aunque al principio se limitaban a los rasgos físicos heredables, fácilmente reconocibles, actualmente hay técnicas más elaboradas que permiten crear mapas de ligamiento comparando la posición de genes diana en comparación con el orden de los marcadores genéticos o de partes conocidas del ADN.
La cartografía física es capaz de medir la distancia real entre puntos de los cromosomas. Las técnicas más avanzadas combinan robótica, informática y uso de láser para calcular la distancia entre marcadores genéticos conocidos. Para conseguirlo, se fragmenta el ADN de los cromosomas humanos aleatoriamente. A continuación se duplican muchas veces para estudiar en los clones, que son las secuencias duplicadas, la ausencia o presencia de marcas genéticas identificables. Los clones que comparten varias marcas provienen de segmentos solapados normalmente. Estas regiones pueden utilizarse después para determinar el orden de las marcas en los cromosomas y su secuencia. Para obtener la secuencia real de nucleótidos hacen falta mapas físicos altamente detallados que recogen el orden de las piezas clonadas con exactitud.
En el Proyecto Genoma Humano se utilizó un método de secuenciación desarrollado por Frederick Sanger, bioquímico británico y dos veces premio Nobel. Este método replica piezas específicas de ADN y las modifica de modo que acaben en una forma fluorescente.
Actualmente se detecta el nucleótido modificado del extremo de las cadenas con modernos secuenciadores de ADN automáticos. Estos determinan los nucleótidos que hay exactamente en la cadena. A continuación se combina esta información de manera informatizada, y así se reconstruye la secuencia de pares de bases del ADN original.
Un aspecto muy importante es duplicar rápidamente y con exactitud el ADN, tanto para después cartografiarlo como para secuenciarlo. Al comienzo de la investigación en este campo se clonaba el material genético introduciéndolo en organismos unicelulares de rápida división, pero en la década de los ochenta se generalizó el uso de la PCR (reacción en cadena de polimerasa). Esta técnica se puede automatizar fácilmente y es capaz de copiar una sola molécula de ADN muchos millones de veces en poco tiempo. Kary Mullis obtuvo el Premio Nobel de Química por idearla, en 1993.
Donantes de genoma
El PGH e IHGSC internacional (sector público) recogieron el semen de hombres y la sangre de mujeres de muchos donantes diferentes, pero solo unas pocas de estas muestras fueron estudiadas después realmente. Así se garantizó que la identidad de los donantes estuviera salvaguardada de modo que nadie supiera qué ADN sería el secuenciado. También han sido utilizados clones de ADN de varias bibliotecas, la mayoría de las cuales fueron creadas por el Dr. J. Pieter de Jong. Se comunicó de manera informal, pero es bien conocido por la comunidad en general, que gran parte del ADN secuenciado provenía de un único donante anónimo de Buffalo, Nueva York, su nombre en clave era RP11. Los científicos encargados utilizaron principalmente los glóbulos blancos de dos hombres y dos mujeres elegidos aleatoriamente.
Ventajas
El trabajo sobre la interpretación de los datos del genoma se encuentra todavía en sus etapas iniciales. Se prevé que un conocimiento detallado del genoma humano ofrecerá nuevas vías para los avances de la medicina y la biotecnología. Por ejemplo, un número de empresas, como Myriad Genetics ha empezado a ofrecer formas sencillas de administrar las pruebas genéticas que pueden mostrar la predisposición a una variedad de enfermedades, incluyendo cáncer de mama, los trastornos de la hemostasia, la fibrosis quística, enfermedades hepáticas y muchas otras. Además, la etiología de los cánceres, la enfermedad de Alzheimer y otras áreas de interés clínico se consideran susceptibles de beneficiarse de la información sobre el genoma y, posiblemente, pueda a largo plazo conducir a avances significativos en su gestión.
Hay también muchos beneficios tangibles para los biólogos. Por ejemplo, un investigador de la investigación de un determinado tipo de cáncer puede haber reducido su búsqueda a un determinado gen. Al visitar la base de datos del genoma humano en la World Wide Web, este investigador puede examinar lo que otros científicos han escrito sobre este gen, incluyendo (potencialmente) la estructura tridimensional de su producto; su/s función/es; sus relaciones evolutivas con otros genes humanos, o genes de ratones, levaduras, moscas de la fruta; las posibles mutaciones perjudiciales; las interacciones con otros genes; los tejidos del cuerpo en el que este gen es activado; las enfermedades asociadas con este gen u otro tipo de datos. Además, la comprensión más profunda de los procesos de la enfermedad en el ámbito de la biología molecular puede determinar nuevos procedimientos terapéuticos. Dada la importancia del ADN en biología molecular y su papel central en la determinación de la operación fundamental de los procesos celulares, es probable que la ampliación de los conocimientos en este ámbito facilite los avances médicos en numerosas áreas de interés clínico que puede no haber sido posible por otros métodos.
El análisis de las similitudes entre las secuencias de ADN de diferentes organismos es también la apertura de nuevas vías en el estudio de la evolución. En muchos casos, las cuestiones de evolución ahora se pueden enmarcar en términos de biología molecular y, de hecho, muchos de los grandes hitos evolutivos (la aparición de los ribosomas y orgánulos, el desarrollo de planes de embriones con el cuerpo, el sistema inmune de vertebrados) pueden estar relacionados a nivel molecular. Muchas de las preguntas acerca de las similitudes y diferencias entre los seres humanos y nuestros parientes más cercanos (los primates, y de hecho los otros mamíferos) se espera que sean iluminados por los datos de este proyecto.
El Proyecto Diversidad del Genoma Humano (PDGH), derivado de investigaciones dirigidas a la asignación del ADN humano – que varía entre los grupos étnicos – que se rumorea que ha sido detenido, realmente continúa y hasta la fecha ha arrojado nuevas conclusiones. En el futuro, el PGH podría exponer nuevos datos en la vigilancia de las enfermedades, el desarrollo humano y la antropología. El PGH podría desbloquear secretos y crear nuevas estrategias para combatir la vulnerabilidad de los grupos étnicos a ciertas enfermedades. También podría mostrar cómo las poblaciones humanas se han adaptado a estas vulnerabilidades.
Además, el PGH tiene una consecuencia muy importante, y es que se pueden conocer la base molecular de ciertas enfermedades hereditarias y que se puede realizar un diagnóstico de las mismas:
Conocer las bases moleculares de las enfermedades hereditarias
Una de las aplicaciones más directas de conocer la secuencia de genes que componen el genoma humano es que se puede conocer la base molecular de muchas enfermedades genéticas y se puede realizar un diagnóstico adecuado. Algunas de estas enfermedades son las siguientes:
Enfermedad de Gaucher: esta enfermedad es producida por una mutación recesiva en el gen que codifica la enzima glucocerebrosidasa, que se localiza en el cromosoma 1. Esta enzima se encarga de metabolizar los glucocerebrósidos (un tipo de lípidos). En los enfermos de Gaucher, estos lípidos no pueden ser descompuestos y se acumulan principalmente en el hígado, en el bazo y en la médula ósea. Los síntomas de la enfermedad de Gaucher incluyen fuertes dolores, fatiga, ictericia, daños óseos, anemia y muerte. Gracias al PGH se pudo realizar la primera terapia efectiva contra esta enfermedad, inyectándose la enzima sintetizada en escherichia coli en el torrente sanguíneo de los enfermos. Esto detiene el avance de los síntomas y en muchos casos los revierte.
Enfermedad de Alzheimer: Esta enfermedad es una enfermedad degenerativa que destruye el cerebro, haciendo que los enfermos pierdan la memoria y el juicio, y que finalmente impide que se puedan valer por sí solos. El único método seguro para diagnosticar la enfermedad de Alzheimer se encuentra en la autopsia, pero actualmente, mediante resultados obtenidos con la resonancia magnética y tomografía por emisión de positrones de las proteínas beta amiloide y tau, los investigadores pueden detectar cambios cerebrales asociados a la fase preclínica (hasta 20 años antes de los primeros síntomas) de la enfermedad. El Alzheimer esporádico es el más común y de origen multifactorial, aunque el mayor factor de riesgo sea la edad, mientras que el Alzheimer de origen genético ronda en un 1% de los casos. Gracias al PGH se han localizado marcadores para el Alzheimer de origen genético en los cromosomas 1, 14, 19 y 21.
Enfermedad de Huntington: Esta enfermedad es también una enfermedad degenerativa y conduce a un deterioro mental que termina en demencia. Normalmente comienza a aparecer entre los 30 y los 50 años y presenta síntomas tales como cambios en la personalidad y en el estado de ánimo, depresión y pérdida gradual del control sobre los movimientos voluntarios, causando espasmos primero y grandes movimientos al azar posteriormente. Esta enfermedad presenta una herencia autosómica dominante, es decir, si uno de los padres la posee, sus hijos tienen el 50% de probabilidad de padecerla también. La Enfermedad de Huntington no se salta generaciones. Si no se hereda el gen, no se puede transmitir a la descendencia. Del mismo, modo, si se hereda el gen, inevitablemente se padecerá la enfermedad, más tarde o más temprano. En 1993 se consiguió aislar el gen que provoca esta enfermedad, localizado en el cromosoma 4, y en lo que se han ido desarrollando las investigaciones posteriores, ha sido fundamentalmente en conocer las razones que hacen que la Enfermedad de Huntingnton se manifieste de forma tardía, y muchas líneas de investigación están dirigidas a encontrar un tratamiento y una cura.
Síndrome de Marfan: Es una enfermedad congénita del tejido conectivo que afecta a numerosos órganos y sistemas, incluyendo el esqueleto, los pulmones, los ojos, el corazón y los vasos sanguíneos. Esta enfermedad se caracteriza por un crecimiento anormal de las extremidades (especialmente de los dedos), una dislocación parcial del cristalino (en el 50% de los pacientes), anormalidades cardiovasculares (la arteria aorta suele ser más ancha y más frágil que en las personas normales) y otras deformaciones. El síndrome de Marfan es también una enfermedad autosómica dominante, por lo que los descendientes de personas afectadas poseen el 50% de posibilidades de padecerla. La enfermedad está asociada al gen FBN1, localizado en el cromosoma 15. El FBN1 codifica una proteína llamada fibrilina, que es esencial para la formación de fibras elásticas del tejido conectivo. Sin el soporte estructural de las fibras elásticas, muchos tejidos presentan una debilidad que puede conducir los síntomas comentados anteriormente.
Gracias al PGH se han podido estudiar y diagnosticar
, de una u otra manera, las secuencias genéticas tras la secuenciación del genoma por el Proyecto Genoma Humano. El diagnóstico de cierta enfermedad, gracias al PGH se puede realizar de manera presintomática y prenatal.
El conocimiento de la base molecular de las enfermedades permite realizar el diagnóstico presintomático y gracias a él tomar medidas preventivas, como alteraciones en el estilo de vida, evitar la exposición a factores de riesgo, realizar un seguimiento continuo del individuo o realizar intervenciones puntuales, para poder tratar la enfermedad aunque todavía no haya aparecido.
En cuanto al diagnóstico prenatal, éste consiste en un conjunto de técnicas que sirven para conocer la adecuada formación y el correcto desarrollo del feto antes de su nacimiento, para poder conocer posibles malformaciones desde los primeros estadios de desarrollo del embrión. La técnica más común de diagnóstico prenatal es la amniocentesis, que consiste en el análisis del líquido amniótico que rodea al feto durante el embarazo. Las células desprendidas del feto y que flotan en dicho líquido sirven para obtener un recuento exacto de cromosomas y para detectar cualquier estructura cromosómica anormal. El diagnóstico prenatal conlleva una importante polémica. Las mujeres cuyo hijo se observe que presentan características de padecer cierta enfermedad o que presentan malformaciones en sus cromosomas, decidirán abortar, lo que para los detractores del aborto es una aberración. La polémica está también alimentada por el hecho de que se pueden conocer tanto enfermedades que se desarrollen desde el primer día de vida del individuo como enfermedades que pueden aparecer a su edad avanzada, como el Alzheimer, por ejemplo. En ese caso, ¿abortaríamos a un feto que puede presentar la Enfermedad de Alzheimer casi al final de su vida, privándole de una vida previa normal? Esto conlleva también a realizar un baremo de qué enfermedades podrían considerarse suficientes para realizar el aborto, poniéndose por ejemplo, el daltonismo.
Por otra parte, y como consecuencia del desarrollo de las técnicas de la fecundación in vitro, hoy en día se puede realizar el conocido como diagnóstico genético preimplantacional (DGPI). Éste permite testar los embriones desde un punto de vista genético y cromosómico para así elegir el que se encuentre sano e implantarlo en el útero de la madre. El DGPI evita la gestación de un niño afectado genética o cromosómicamente, y conlleva la decisión de los padres de realizar, en su caso, un aborto terapéutico.
Terapia génica, terapia farmacológica y medicina predictiva]
Una vez que se conocen qué genes producen qué enfermedades, y las características para diagnosticar una enfermedad conociendo la secuencia de bases, es necesario realizar una terapia para acabar con esa enfermedad, ya que de ser de otra manera, el diagnóstico de una enfermedad no es más que una carga emocional que el paciente tiene que soportar de la mejor manera posible, conviviendo con la impotencia y la ansiedad que le puede suponer a un paciente el saber que en un determinado lapso de tiempo es posible que padezca una enfermedad. Una consecuencia, por tanto, del PGH es desarrollar terapias contra las enfermedades que ha diagnosticado. Se conocen la terapia génica, la terapia farmacológica y la medicina predictiva:
La terapia génica es una consecuencia directa del PGH y supone la probabilidad de curar las enfermedades hereditarias cartografiadas por éste, insertando copias funcionales de genes defectivos o ausentes en el genoma de un individuo para tratar dicha enfermedad. Las técnicas actuales de terapia génica no pueden asegurar que el gen se inserte en un lugar apropiado del genoma, existe la posibilidad de que interfiera con el funcionamiento de un gen importante o incluso que active un oncogén, provocando así un cáncer en el paciente. Sin embargo, estas técnicas sólo se utilizan con pacientes que ya corren peligro inminente de muerte, por lo que la posibilidad de contraer un cáncer en un futuro incierto no constituye un impedimento muy grave para aceptar el tratamiento.
El primer caso que se conoce de terapia génica tuvo lugar en los NIH (National Institutes of Health. En español: Institutos Nacionales de la Salud), en Bethesda, Maryland. Consistió en la inoculación de glóbulos blancos genéticamente modificados a una niña que padecía inmunodeficiencia severa combinada (deficiencia de adenosina-desaminasa o ADA). Esta enfermedad es una enfermedad rara, y la carencia de ADA se puede tratar con trasplantes de médula ósea. Sin embargo, el trasplante sólo es posible si el paciente tiene un hermano que no esté afectado por la enfermedad y que sea compatible. Otra posibilidad es inyectar la proteína directamente, pero las inyecciones no llegan inmediatamente al lugar necesario y constituyen un mal sucedáneo de los sutiles mecanismos que controlan y dirigen la producción de ADA en circunstancias normales. La operación consistió en la extracción de linfocitos T de la paciente, su modificación genética y su reimplantación. Con esto las células comenzaron a producir la ADA.
Cuando se realizó esta primera intervención, los doctores de los NIH estudiaron las implicaciones éticas que podía tener esta operación y llegaron a la conclusión de que no existía diferencia moral con respecto a cualquier tipo de trasplante de tejidos o de órganos. Esta comparación residía en que los genes trasplantados sólo afectaban a las células somáticas del individuo, de modo que sólo afectaban a la niña misma y que no lo harían por tanto a su descendencia. Podemos diferenciar entonces dos tipos de terapia génica, en línea somática y en línea germinal. Esta última consiste en introducir genes nuevos, biológicamente funcionales, en células germinales (óvulos y/o espermatozoides) antes de que se produzca la fecundación. El embrión que surge tras la fecundación partirá de una única célula modificada genéticamente, por lo que todas sus células posteriores presentarán la misma modificación, incluyendo las futuras células germinales que producirá, pudiendo transmitir sus características a las generaciones futuras.
Todos los estudios nacionales han rechazado la terapia en línea germinal, de momento, ya que opinan que todavía no se dispone de los suficientes conocimientos para evaluar los riesgos que supone este tipo de terapia y que es necesario realizar un estricto examen ético antes de comenzar a aplicarla, si esto se acabara produciendo.
La terapia farmacológica se ve también facilitada por el PGH ya que éste permite encontrar alteraciones en la secuencia del ADN de genes específicos y esto conlleva a que se realice el tratamiento con medicamentos de una manera dirigida, neutralizando las alteraciones y modificando favorablemente el curso de la enfermedad de forma más efectiva que los tratamientos de la medicina actual, que están generalmente dirigidos a aliviar los síntomas.
El PGH permite además, en relación con la farmacología, modificar los medicamentos para que se ajusten a las características genéticas del paciente y así poder metabolizar el fármaco de la mejor manera posible, lo que en consecuencia, elimina o minimiza los efectos secundarios indeseables del mismo. Gracias al PGH el médico tendrá un perfil genético del paciente antes de iniciar el tratamiento.
La medicina predictiva permite diagnosticar enfermedades, gracias a los conocimientos del genoma, que aún no se han desarrollado en el paciente. Se distinguen dos tipos de enfermedades que se pueden diagnosticar mediante la medicina predictiva. Las monogénicas, que se pueden identificar fácilmente ya que se conocen perfectamente las leyes deterministas que las regulan; y las poligénicas, para cuyo buen estudio es necesario realizar sondeos poblacionales. Por ejemplo se pueden encontrar los genes que regulan el nivel de colesterol en la sangre (unos veinte). Determinadas combinaciones de variedades de estos genes sitúan al sujeto en un grupo de riesgo de padecer enfermedades tempranas de las arterias coronarias y ataques cardíacos. Si además el sujeto lleva una dieta rica en grasas animales y una vida sedentaria (también influyen por tanto agentes externos como puede ser el modo de vida y la alimentación), es muy posible que muera de infarto antes de los cincuenta años. La meta es conocer exactamente qué combinaciones de genes son especialmente peligrosas y en esto tiene un papel muy importante el Proyecto Genoma Humano. La medicina predictiva también causa una importante controversia en la sociedad ya que los estudios poblaciones que se realizan para estudiar las enfermedades poligénicas se pueden utilizar para discriminar a ciertas personas o grupos, lo que se llamaría discriminación genética. Este tema se tratará en el apartado Aspectos Éticos.
Aspectos éticos y controversia
Aunque la medicina proporciona la base para la evolución de la bioética, actualmente somos testigos de su aplicación a la investigación científica relacionada. Así pues, el PGH ha dado lugar a una de las áreas de conocimiento biológico con mayor crecimiento. Los conocimientos genómicos derivados del Proyecto Genoma Humano, se utilizan para mejores y más rápidos diagnósticos basados en el análisis directo del ADN, e incluso para el diagnóstico prenatal en aquellos casos en los que se sospecha que el bebé tenga alteraciones morfológicas, funcionales o ponga en peligro la vida de su madre. También es posible aplicar este conocimiento a personas asintomáticas para averiguar si han heredado de algún progenitor una mutación causal de una enfermedad genética que pueda desarrollarse en el futuro.
Así planteado el tema, se percibe entonces una importante brecha entre la capacidad diagnóstica y predictiva del conocimiento genómico por un lado, y la falta de intervenciones preventivas y terapéuticas por otro, lo que lleva a conflictos éticos surgidos del Proyecto Genoma Humano. Además hay determinadas áreas como el asesoramiento a parejas en riesgo de transmitir enfermedades genéticas a su descendencia, que han suscitado mucho interés y para las que se han dictado una serie de principios éticos:
Respeto a la dignidad individual y a la inteligencia básica de las personas, así como a sus decisiones médicas y reproductivas (libre elección de interrumpir o continuar un embarazo con riesgo).
Informar objetivamente al paciente sin tener en cuenta los valores subjetivos del profesional médico.
Protección a la privacidad de la información genética.
Desmitificación del Proyecto Genoma Humano, aclarando verdaderamente su alcance con acciones específicas en educación.
Otro problema de gran importancia es la obtención de patentes de genes por parte de compañías biotecnológicas, gobiernos y centros de investigación universitarios, para una posterior venta o explotación comercial, sin tener en cuenta que parte de los fondos empleados en el PGH era de los contribuyentes. También debemos observar el PGH contextualizado social e históricamente, atendiendo a la desigualdad social y económica entre países, que va a producir una inequidad en el acceso a los beneficios que se extraigan de la investigación.
Una solución a todas estas tensiones podría ser la formación de profesores de ciencias o la enseñanza directa a estudiantes como una forma de abrir las mentes y aclarar definitivamente el alcance del Proyecto Genoma Humano en la sociedad. Pero es imprescindible incorporar temas de bioética a los programas de enseñanza.
Tanto en Estados Unidos como en la Unión Europea se han desarrollado programas para contemplar las consecuencias éticas y sociales de la investigación científica y que no se produzcan conflictos. En Estados Unidos se encuentra el ELSI y fuera de ellos se encuentra la Declaración Universal sobre el Genoma Humano y los Derechos Humanos, promovida por la UNESCO
El ELSI es el Programa Ético, Legal y Social (Ethical, Legal and Social Implications Research Program, en inglés) que desarrolló el NHGRI (National Humane Genome Research Institute, en inglés, o Instituto Nacional de Investigación del Genoma Humano, de Estados Unidos) en 1990. Este programa permite un acercamiento a la investigación científica teniendo en cuenta las implicaciones éticas, legales y sociales que ésta supone, al mismo tiempo que se está investigando para, de esta manera, poder identificar los posibles futuros problemas y solucionarlos antes de que la información científica se extienda. El programa de investigación ELSI tiene un papel muy importante en todo lo relacionado con el PGH, y se encarga de analizar las implicaciones éticas y sociales de la investigación genética de la siguiente manera:
Examinando las ediciones que rodean la terminación de la secuencia humana del ADN y del estudio de la variación genética humana.
Examinando las ediciones llevadas a cabo por la integración de tecnologías e información genética para el cuidado médico y actividades de la salud pública.
Explorando las maneras en las cuales el nuevo conocimiento genético puede actuar recíprocamente con una variedad de perspectivas éticas, filosóficas y teológicas.
Explorando cómo influyen en el uso e interpretación de la información genética, de la utilización de servicios genéticos y del desarrollo de la política, los factores y los conceptos socioeconómicos de la raza y de la pertenencia étnica.
Para alcanzar estas metas, las actividades y la investigación del programa de ELSI se centran en cuatro áreas del programa:
Aislamiento e imparcialidad en el uso y la interpretación de la información genética.
Integración clínica de las nuevas tecnologías genéticas.
Ediciones que rodean la investigación de la genética.
Educación pública profesional.
El ELSI también ha iniciado una serie de emprendimientos educacionales que están dirigidos a entrenar a profesionales de la salud para que puedan interpretar los nuevos tests diagnósticos basados en el ADN que comenzarán a surgir más y más frecuentemente gracias a la información obtenida del PGH. Además de esta formación de profesionales de la salud también se necesita que los políticos y el público en general tengan un criterio suficiente sobre algunos asuntos críticos relacionados con las pruebas genéticas. Por ello, es necesario extender la información genética en las escuelas, los medios de comunicación, alentar la discusión pública sobre el tema y suministrar también información a los políticos. Una de las iniciativas es el establecimiento de la Coalición Nacional para la Educación de los Profesionales de la Salud en Genética (NCHPEG), también en EE. UU., pero rápidamente se queda insuficiente ya que sólo abarca a los profesionales de la Salud.
Declaración Universal sobre el Genoma Humano y los Derechos Humanos]
Así como Estados Unidos tiene un programa para regular las implicaciones sociales y éticas que tienen las investigaciones científicas para tratar de regularlas y que no haya conflictos, la UNESCO redactó en 1997 la “Declaración Universal sobre el Genoma Humano y los Derechos Humanos”, cuyo prefacio es el siguiente:
La Declaración Universal sobre el Genoma Humano y los Derechos Humanos, aprobada el 11 de noviembre de 1997 por la Conferencia General en su 29ª reunión por unanimidad y por aclamación, constituye el primer instrumento universal en el campo de la biología. El mérito indiscutible de ese texto radica en el equilibrio que establece entre la garantía del respeto de los derechos y las libertades fundamentales, y la necesidad de garantizar la libertad de la investigación. La Conferencia General de la UNESCO acompañó esa Declaración de una resolución de aplicación, en la que pide a los Estados Miembros que tomen las medidas apropiadas para promover los principios enunciados en ella y favorecer su aplicación. El compromiso moral contraído por los Estados al adoptar la Declaración Universal sobre el Genoma Humano y los Derechos Humanos es un punto de partida: anuncia una toma de conciencia mundial de la necesidad de una reflexión ética sobre las ciencias y las tecnologías. Incumbe ahora a los Estados dar vida a la Declaración con las medidas que decidan adoptar, garantizándole así su perennidad.
Federico Mayor, 3 de diciembre de 1997.
Está compuesta por 25 artículos que se dividen en las siguientes áreas, destacando en cada una de ellas un determinado artículo:
La dignidad humana y el genoma humano. Contiene los 4 primeros artículos y establece la base la declaración y su objeto, el ser humano y el genoma humano. Cabe destacar el artículo 1: “El genoma humano es la base de la unidad fundamental de todos los miembros de la familia humana y del reconocimiento de su dignidad intrínseca y su diversidad. En sentido simbólico, el genoma humano es el patrimonio de la humanidad”.
Derechos de las personas interesadas. Está compuesta por los artículos desde el 5 al 9 y presenta los derechos que tienen las personas como portadoras de los genes y sus consecuencias sociales. Cabe destacar el artículo 6 porque está relacionado con la discriminación genética, que será tratada más adelante: “Nadie podrá ser objeto de discriminaciones fundadas en sus características genéticas, cuyo objeto o efecto sería atentar contra sus derechos humanos y libertades fundamentales y el reconocimiento de su dignidad”.
Investigaciones sobre el genoma humano. Formada por los artículos 10, 11 y 12. Trata la imposición de la dignidad humana sobre cualquier tipo de investigación relativa al genoma humana, el derecho de todas las personas a acceder a los progresos de la biología y a la orientación de la investigación en el campo de la biología, genética y medicina hacia un alivio del sufrimiento y una mejora de la salud del individuo y de toda la humanidad. Se puede destacar el artículo 10 que alienta a los Estados miembros a actuar sobre posibles conductas contrarias a la declaración: “No deben permitirse las prácticas que sean contrarias a la dignidad humana, como la clonación con fines de reproducción de seres humanos. Se invita a los Estados y a las organizaciones internacionales competentes a que cooperen para identificar estas prácticas y a que adopten en el plano nacional o internacional las medidas que correspondan, para asegurarse de que se respetan los principios enunciados en la presente Declaración”.
Condiciones de ejercicio de la actividad científica. Contiene los artículos del 13 al 16 y en ellos se otorga a los Estados miembros la potestad de regular las actividades relacionadas con la investigación y de crear organismos para regular las consecuencias éticas y sociales causadas por ella, como declarar el artículo 16: “Los Estados reconocerán el interés de promover, en los distintos niveles apropiados, la creación de comités de ética independientes, pluridisciplinarios y pluralistas, encargados de apreciar las cuestiones éticas, jurídicas y sociales planteadas por las investigaciones sobre el genoma humano y sus aplicaciones”.
Solidaridad y cooperación internacional. Esta parte está formada por los artículos 17, 18 y 19 y se refiere a la cooperación y solidaridad tanto entre los individuos que forman los Estados miembros como entre los Estados mismos, refiriéndose en primer lugar a casos como enfermedades genéticas y en el segundo a compartir conocimientos científicos sobre el genoma humano entre países que tengan una gran investigación desarrollada y otros que la tengan menos, como dice el artículo 18: “Los Estados deberán hacer todo lo posible, teniendo debidamente en cuenta los principios establecidos en la presente Declaración, para seguir fomentando la difusión internacional de los conocimientos científicos sobre el genoma humano, la diversidad humana y la investigación genética, y a este respecto favorecerán la cooperación científica y cultural, en particular entre países industrializados y países en desarrollo”.
Fomento de los principios de la Declaración. Son los artículos 20 y 21 e impulsan a los Estados miembros de la UNESCO a fomentar y extender los principios entre los individuos que los forman, también entre los políticos, y además comprometerse a favorecer el debate abierto y la libre expresión de corrientes socioculturales, religiosas o filosóficas. El artículo 20 también impulsa la información desde la educación: “Los Estados tomarán las medidas adecuadas para fomentar los principios establecidos en la Declaración, a través de la educación y otros medios pertinentes, y en particular, entre otras cosas, la investigación y formación en campos interdisciplinarios y el fomento de la educación en materia de bioética, en todos los niveles, particularmente para los responsables de las políticas científicas”.
Aplicación de la Declaración. Los artículos del 22 al 25 se refieren a la obligación de los Estados de fomentar el respeto frente a los enunciados de la Declaración, difundirlos y hacerse cargo de que se realicen correctamente. Así, el artículo 23 declara: “Los Estados tomarán las medidas adecuadas para fomentar mediante la educación, la formación y la información, el respeto de los principios antes enunciados y favorecer su reconocimiento y su aplicación efectiva. Los Estados deberán fomentar también los intercambios y las redes entre comités de ética independientes, según se establezcan, para favorecer su plena colaboración”.
Discriminación genética y patente de genes
Entramos ahora en los que posiblemente sean los dos puntos más importantes de la controversia causada por el PGH, que se pasan a explicar a continuación:
Discriminación genética
El ELSI tiene un papel muy importante en el campo de la discriminación genética. Cuando se dieron los primeros pasos del PGH, los científicos tuvieron muy claro desde el principio que era necesario realizar un estudio ético y social, inicialmente a pequeña escala y si era necesario, a mayor; sobre alguna enfermedad que pudiera tener lugar en la sociedad, para evitar cualquier tipo de discriminación genética. Un ejemplo interesante de discriminación genética tuvo lugar en Estados Unidos durante los años setenta y relacionada con una campaña que realizó el gobierno para detectar portadores del gen de la anemia de células falciformes.
Capilares sanguíneos en los que se pueden observar eritrocitos falciformes.
La anemia de células falciformes, además, tiene un componente relacionado con la raza muy importante, ya que es la enfermedad genética más frecuente entre la población negra. Se trata de una enfermedad recesiva bastante cruel ya que los que la sufren no pueden realizar esfuerzos, ya que corren un grave riesgo de sufrir una insuficiencia respiratoria aguda que les ocasione repentinamente la muerte. Pues bien, la discriminación genética aparece cuando el gobierno realizó un estudio poblacional para detectar individuos que portaran este gen. La anemia de células falciformes no tiene cura y por tanto, si alguien era diagnosticado de anemia de células falciformes no poseía la más mínima esperanza de curación. El problema se hizo patente cuando el gobierno declaró obligatorio en varios estados realizar la prueba de detección a los recién nacidos y a los escolares, sin seguir un programa paralelo de orientación genética que pudiera ofrecer consejo a las familias afectadas, y cuando el público comenzó a confundir a las personas portadoras (heterocigóticas) con las enfermas, debido a la completa falta de una campaña informativa. Por si esto fuera poco, Linus Pauling, que había descubierto el método de análisis de la hemoglobina, realizó unas desafortunadas declaraciones en las que sugería que se marcara de alguna manera a los portadores para que no se mezclaran y no tuvieran hijos entre sí. La información que se recogió en este estudio pasó a formar parte del historial médico de los niños que estaban afectados. Las compañías de seguros comenzaron entonces a negarse a formalizar el seguro si conocían que su posible cliente padecía anemia de células falciformes, e incluso si era simplemente portador del gen. También el mercado de trabajo comenzó a discriminar a los enfermos y portadores. A las personas de color que portaban el gen se les negaba por ejemplo el trabajo en compañías aéreas porque se pensaba que su sangre reaccionaría mal al encontrarse a bajas presiones causados por la altura del avión (algo que es erróneo).
Un gran problema que tuvo el caso de la anemia de células falciformes en los años setenta fue que no se conocían métodos de estudio del feto y que tampoco estaba permitido el aborto. Esto se ha podido superar actualmente y es un problema menor para el programa ELSI, ya que ahora sí existe la posibilidad de detectar la enfermedad en el feto y, además de que ya está permitido, el aborto terapéutico tiene una aceptación social casi mayoritaria.
En definitiva, es necesario realizar un estudio social y ético y dar la información necesaria a la opinión pública para que no se produzcan casos de discriminación genética, si ya no tan llamativos como el de la anemia de células falciformes en EE. UU., pero sí a menor escala como puede ser la predisposición hacia enfermedades cardíacas o a las discapacidades mentales, por ejemplo.
Patente de genes
El concepto de «patente de genes» aparece también con la secuenciación del genoma producida por el PGH. Y es que resulta necesario compatibilizar las expectativas terapéuticas y de avance científico con las expectativas de aspecto económico, procurando encontrar un equilibrio razonable entre el altruismo que unos buscan en el conocimiento público de la información proporcionada por el PGH y otros que encuentran esta información suficiente para sacarle provecho económico. Es necesario combinar la moralidad con el interés económico. El elemento fundamental de todo esto se encuentra en las empresas privadas que realizan investigaciones en el genoma humano. Como tales empresas privadas, necesitan obtener un beneficio que supla las grandes inversiones que hacen en investigaciones para obtener posteriormente productos farmacéuticos, desarrollar terapias clínicas u otras aplicaciones. Para esto, necesitan proteger sus hallazgos para que nadie se aproveche de su esfuerzo. La cuestión reside en determinar cuál es el marco jurídico apropiado para garantizar debidamente esas expectativas de beneficio. Es, por tanto, lógico que se tratara de amparar bajo la protección de las patentes a los descubrimientos relacionados con la descodificación y aislamiento del ADN, considerándolo una sustancia o estructura que, como otras, se encuentra en la naturaleza y de cuyo conocimiento se puede derivar algún uso diagnóstico y con el fin de compensar las inversiones económicas realizadas. De este modo, los investigadores o instituciones que patentaran la secuencia parcial o total de cierto gen podrían ser acreedores de los derechos que se derivaran de ella para la obtención de fármacos. Por otro lado, hay gente que piensa que las patentes no hacen más que impedir el desarrollo biotecnológico y que la información que se encuentra en los genes debería ser de acceso público.
Las patentes sobre secuencias totales o parciales de genes continúan estando en una importante controversia y se pueden encontrar tres posiciones diferentes:
La postura de la UNESCO: afirma que el Genoma Humano es patrimonio de la Humanidad y que debe quedar excluido de cualquier apropiación pública o privada.
La postura estadounidense: representada por los NIH y Craig Venter (dueño de la empresa Celera Genomics, empresa biotecnológica involucrada en el estudio del Genoma Humano). Parten de que los genes, por muy esenciales que sean para la vida, no son vida humana, y tampoco pueden clasificarse como materia exclusivamente humana ya que los compartimos con otras especies. Opinan que no hay nada que choque contra los criterios de patentabilidad impuestos por la USPTO (http://en.wikipedia.org/wiki/USPTO_registration_examination), por lo que nada debería impedirles proteger la información obtenida y conseguir beneficios para poder avanzar en sus investigaciones.
La postura europea: se encuentra en una posición intermedia. Niega la patentabilidad de cualquier genoma individual completo pero admite que se puedan patentar los genes humanos individualmente si han sido aislados. También mantiene cláusulas de moralidad que permitan rechazar administrativa o jurisdiccionalmente determinadas solicitudes de patente. (Directiva Europea 98/44/CE Art. 5 https://web.archive.org/web/20160304212509/http://www.cgcom.org/sites/default/files/54_Directiva_98_44_CE.pdf). La Directiva europea pretende solucionar los problemas de las patentes estableciendo una distinción de planos. Por un lado se encontrarían los genes “tal y como se encuentran en la naturaleza”, que actuarían como patrimonio común de la humanidad y a los que se debe proteger, y por otro lado se encontrarían los genes “que han sido aislados de su medio natural por procedimientos técnicos”, sobre los que sí podría implantarse una patente al haberse modificado su naturaleza a través del procedimiento técnico.
Y ahora qué: el proyecto genoma humano[editar]
.
Cifras y datos
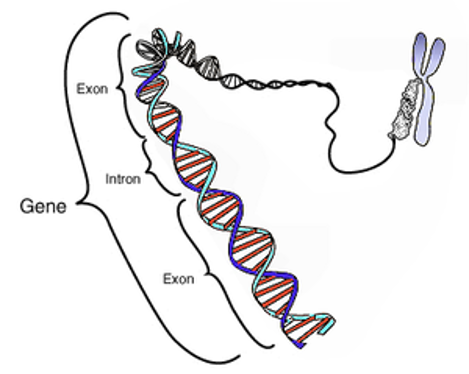
Este diagrama esquemático muestra un gen en relación a su estructura física (doble hélice de ADN) y a un cromosoma (derecha). Los intrones son regiones frecuentemente encontradas en los genes de eucariotas, que se transcriben, pero son eliminadas en el procesamiento del ARN (ayuste) para producir un ARNm formado sólo por exones, encargados de traducir una proteína. Este diagrama es en exceso simplificado ya que muestra un gen compuesto por unos 40 pares de bases cuando en realidad su tamaño medio es de 20 000-30 000 pares de bases).
El Consorcio Internacional, integrado por 20 grupos de diferentes países y por otro lado la empresa privada Celera, hicieron público, el 12 de febrero de 2001, el mapa provisional del genoma humano (GH) que aporta una extraordinaria información acerca de las bases genéticas del ser humano.
El Consorcio Internacional ha calculado que el genoma humano contiene 20 500 genes.
De los 300 000 clones de partida fueron válidos 30 000 clones que representan un total de 3200 megabases. Estos resultados alcanzados en octubre del 2000, representan el 90% del genoma. La secuencia obtenida es de enorme trascendencia y son muchos y variados los puntos de interés pudiendo destacarse algunos datos:
El humano tiene solo el doble de genes que la mosca del vinagre, un tercio más que el gusano común y apenas 5.000 genes más que la planta Arabidopsis.
3200 millones de pares de bases forman genes, repartidos entre los 23 pares de cromosomas. Los cromosomas más densos (con más genes codificadores de proteínas) son el 17, 19 y el 22. Los cromosomas X, Y, 4, 18 y 13 son los más áridos.
El equipo de Celera Genomics utilizó para secuenciar el genoma humano muestras de ADN de tres mujeres y dos hombres (un afroamericano, un chino, un asiático, un hispanomexicano y un caucasiano). El equipo de Celera utilizó ADN perteneciente a doce personas. Cada persona comparte un 99,99 por ciento del mismo código genético con el resto de los seres humanos. Sólo 1250 nucleótidos separan una persona de otra.
Hasta ahora se han encontrado 223 genes humanos que resultan similares a los genes bacterianos.
Sólo un 5 % del genoma codifica proteínas. El 25% del genoma humano está casi desierto, existiendo largos espacios libres entre un gen y otro.
Se calcula que existen entre 250 000 y 300 000 proteínas distintas. Por tanto cada gen podría estar implicado por término medio en la síntesis de unas diez proteínas.
Algo más del 35% del genoma contiene secuencias repetidas. Lo que se conoce como ADN basura.
Se han identificado un número muy elevado de pequeñas variaciones en los genes que se conocen como polimorfismos nucleótidos únicos, SNP de su acrónimo inglés. Celera ha encontrado 2,1 millones de SNP en el genoma y el Consorcio 1,4 millones. La mayoría de estos polimorfismos no tienen un efecto clínico concreto pero de ellos depende, por ejemplo, el que una persona sea sensible o no a un determinado fármaco y la predisposición a sufrir una determinada enfermedad.
Referencias
↑ «BBC NEWS». 14 de abril de 2003. Consultado el 22 de julio de 2006. Texto « Human genome finally complete » ignorado (ayuda); Texto « Science/Nature » ignorado (ayuda)
↑ Baquero F; Nombela C, (18 de julio de 2012). «The microbiome as a human organ». The microbiome as a human organ. PMID 22647038. doi:10.1111/j.1469-0691.2012.03916. Detalles bibliográficos de Proyecto Genoma Humano
Página: Proyecto Genoma Humano
Autor: colaboradores de Wikipedia
Editor: Wikipedia, La enciclopedia libre.
Última revisión: 6 de febrero del 2020, 10:44 UTC
Fecha de consulta: 10 de febrero del 2020, 18:49 UTC
URL permanente: https://es.wikipedia.org/w/index.php?title=Proyecto_Genoma_Humano&oldid=123344887
Código de versión de la página: 123344887
 Telómero básico
Telómero básico
 EL MOVIMIENTO Y LA MEZCLA EN LA EVOLUCIÓN DEL HOMO
EL MOVIMIENTO Y LA MEZCLA EN LA EVOLUCIÓN DEL HOMO
 DIGITALIZACION DE LA GENOMICA
DIGITALIZACION DE LA GENOMICA