La aterosclerosis es un fenómeno patológico focal que afecta a las grandes arterias, principalmente la aorta y las coronarias, carótidas, ilíacas y femorales. El desarrollo de la afección vascular se caracteriza por un comienzo temprano denominado fatty streak o estría grasa. Con el paso de los años este proceso, que es considerado reversible, incrementa su acumulación lipídica y en la adolescencia ya se presentan las primeras lesiones fibrosas. En los años siguientes, estas placas se agrandan y modifican, y en la mayoría de los casos todos estos procesos cursan asintomáticos: la ulceración de la placa, su rotura y trombosis son lo que precipita el evento clínico. Por último, actualmente existe una controversia sobre la clasificación, estandarización y correlación clínica de las placas. Esperamos que los conceptos de este artículo proporcionen al lector los conocimientos y las definiciones objetivas que permitan una mejor comprensión de la implicación de los factores de riesgo, el desarrollo de arteriosclerosis y sus manifestaciones clínicas
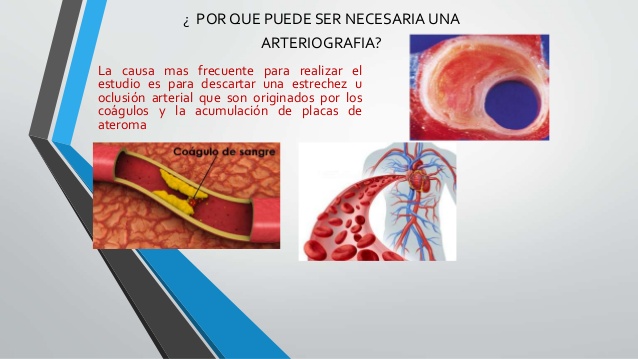
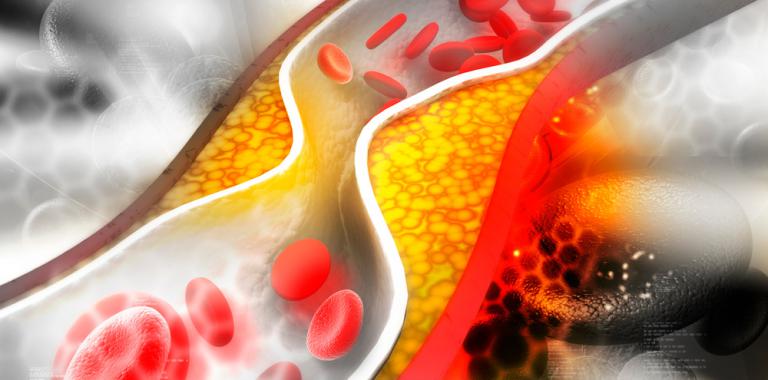
La característica fundamental de esta enfermedad es el depósito de material en la luz de la arteria
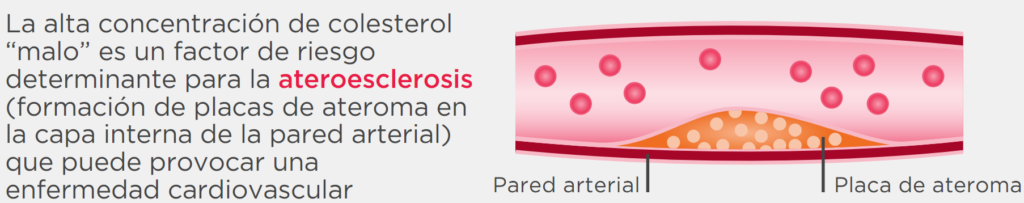
Una placa de ateroma es un cúmulo de colesterol en la pared de una arteria.
Esto se genera porque en condiciones en que el colesterol en la sangre está elevado este hace que la pared de los vasos sanguíneos sea permeable a este colesterol y se acumule en el interior de las arterias.
Junto con el colesterol van a pasar al interior de las arterias leucocitos, en concreto los monocitos.
Ya dentro de la pared van a comerse el colesterol y se genera una célula muy dañina para la pared vascular que se llama célula espumosa. Que son monocitos cargados de colesterol. muy inflamatorias y producen también muchos radicales libres. Eso va a poder en algún momento facilitar que la placa de ateroma se pueda romper y facilitar que las plaquetas se activen y que se forme un trombo.
Las placas de ateroma más peligrosas son las más pequeñas porque son las más inestables.
.
Historia natural de la arteriosclerosis.
Las arterias sufren un proceso natural a lo largo de la vida que se caracteriza por incremento en el espesor del área de la íntima, pérdida de elasticidad, aumento del contenido en calcio y modificaciones en su diámetro; estos cambios ocurren en el sistema arterial principal y se conocen con el nombre genérico de arteriosclerosis. En contraste con este proceso natural, la aterosclerosis es un fenómeno patológico focal que afecta a las grandes arterias, principalmente la aorta y las coronarias, carótidas, ilíacas y femorales. En el año 1958, un estudio realizado por la OMS definió la aterosclerosis como una combinación variable de cambios en la íntima de las arterias consistente en una acumulación focal de lípidos, hidratos de carbono complejos, sangre y productos sanguíneos, tejido fibroso y depósito de calcio, que se asocia con cambios en la íntima media arterial2. Una de las últimas teorías considera que la aterosclerosis es una respuesta inflamatoria y fibroproliferativa excesiva, que se cronifica y no ejerce efecto protector de una serie de agresiones de la íntima arterial que favorecen el depósito de lípidos, lo que influye en la progresión de la placa3.
Las lesiones arterioscleróticas resultan de una variedad de procesos patogénicos que incluye la formación de macrófagos espumosos y muerte y acumulación de lípido extracelular, desplazamiento y reducción de la matriz intercelular estructural y las células musculares lisas, la generación de depósitos minerales, la inflamación crónica, neovascularización, rotura de la superficie de la lesión y formación o transformación de hematoma o trombo en tejido fibromuscular.
A principios de siglo, fueron identificados dos tipos de lesión que se asociaron con la aterosclerosis: la denominada por Rockitansky estría grasa y la placa fibrosa descrita por Virchow4. Sin embargo, estos dos tipos de lesiones no fueron universalmente aceptados como expresión temprana y avanzada de una misma enfermedad. Ludwig Aschoff reconoció en 1924 dos componentes en la enfermedad, uno lipídico en jóvenes (aterosis o ateromatosis) y otro escleroso, fibrolipídico en la edad adulta, llamado aterosclerosis5-7. De hecho el autor habla de tres estados de desarrollo: aterosis en niños, aterosis en adolescentes y aterosclerosis en adultos, que constituyen las lesiones fibrolipídicas (placas fibrosas).
Los conocimientos actuales comienzan a mediados del siglo xx con los distintos estudios anatomopatológicos llevados a cabo en poblaciones de jóvenes fallecidos de forma violenta, realizados con el propósito de estimar la prevalencia de lesiones, y que han supuesto un avance en el estudio del desarrollo de la lesión aterosclerótica. Estos trabajos se realizaban observando las lesiones en la íntima arterial con la sola ayuda de la vista una vez abiertas longitudinalmente, lo que permitía hacer una rápida estimación del porcentaje de íntima arterial con lesiones ateroscleróticas, y la descripción o clasificación se hacía básicamente con los términos utilizados por Aschoff, además de introducir algunos términos empleados por otros autores. Dentro de este contexto podemos referirnos a los trabajos de Enos de los años cincuenta8 en los que da un sustrato anatómico a la hipótesis de que la aterosclerosis comienza en la infancia y se desarrolla en las décadas siguientes, objetivando en jóvenes soldados fallecidos en la guerra de Corea la existencia de lesiones arterioscleróticas avanzadas (en el 77% de los corazones existía alguna evidencia de arteriosclerosis coronaria, con lesiones que iban desde el mero engrosamiento fibroso hasta la oclusión total). Algunos grupos de investigadores9,10 describen una clasificación que consiste en la secuencia de estría grasa, placa fibrosa y lesión complicada (por hemorragia, fisura o ulceración y desarrollo de hematoma o trombo). Además de estos tres términos mencionados, la clasificación de la Organización Mundial de la Salud (OMS)11 de los años cincuenta incluye el término ateroma, que supone lesiones avanzadas con un componente predominantemente lipídico para diferenciarla de aquellas con un componente predominantemente colágeno, que serían las placas fibrosas. La secuencia de Aschoff y las clasificaciones establecidas en los años cincuenta, incluida la de la OMS, fueron aceptadas, y esta organización dirigió a varios grupos de expertos para investigar las características de la lesión aterosclerótica. En 1958 uno de estos grupos dirigidos por Holman10 examinó las aortas de 256 individuos jóvenes de Nueva Orleans y elaboraron un informe al Comité de Lesiones de la Sociedad Americana para el Estudio de la Aterosclerosis, señalando que la calcificación de cualquier lesión aterosclerótica era uno de los procesos que contribuían a su complicación (placa), y estos mismos autores consideraron que las placas calcificadas y las complicadas comportaban igual gravedad, por lo que las llamaron placa complicada. Esta clasificación fue utilizada en el Proyecto Internacional de Arteriosclerosis (IAP) y distinguía entre estría grasa, placa fibrosa y la lesión calcificada o complicada con hemorragia, ulceración o trombosis (tabla 1). En 1969 Strong y McGuill continuaron el estudio de Nueva Orleans e incluyeron las coronarias, detectando placas fibrosas en la segunda década de la vida. Durante los años sesenta y setenta, científicos del IAP que estudiaron autopsias de diferentes orígenes geográficos encontraron estrías grasas en la aorta como un fenómeno universalmente presente en la juventud.
Haimovici12 consideró en 1977 que había un estadio preclínico (estría y placa fibrosa no obstructiva) y dos estadios clínicos, el de aterosclerosis sintomática y el de necrosis de diversos órganos. En 1986 Ross resumió los grandes avances sobre la patogenia y planteó la hipótesis denominada monoclonal13.
En los años ochenta un grupo de científicos inició un estudio para investigar la asociación de factores de riesgo de enfermedad coronaria con la aterosclerosis en adolescentes y adultos jóvenes, estudio conocido con las siglas PDAY (Pathological Determinants Atherosclerosis in Youth), que agrupó 14 laboratorios de los EE.UU. y ha aportado datos muy valiosos en este campo.
En 1992 Fuster propuso una clasificación basada en la patofisiología, con tres tipos de lesiones. A partir de ese año y a través de varias publicaciones, Stary propone una nueva clasificación de la lesión aterosclerótica en seis tipos, ampliada posteriormente a ocho, basándose en la microscopia y en datos obtenidos de arterias coronarias y aortas procedentes de 1.286 autopsias1
Las lesiones de aterosclerosis son el resultado de una variedad de procesos patogénicos, desde la formación de la célula espumosa del macrófago, la apoptosis celular, la acumulación del lípido celular, el desplazamiento y la reducción de la matriz intercelular estructural y la proliferación de las células del músculo liso, los depósitos minerales, la inflamación crónica, la neovascularización, las roturas de la lesión y la formación y transformación de hematoma y trombosis, y por último al tejido fibromuscular. Estos procesos varían en su evolución. En las fases avanzadas de enfermedad, muchos de los procesos pueden correr al unísono y otros no.
La progresión de la arteriosclerosis coronaria describe la morfología de la lesión y fases en relación con las manifestaciones clínicas. El color amarillo indica la acumulación lipídica, el rojo la trombosis y la hemorragia y, por último, el verde la calcificación y el tejido fibroso. Los números romanos indican los tipos de lesión. Las fases 1 y 2 son asintomáticas: la fase 1 con lesiones tempranas I-III, que evolucionan en una fase 2 a lesiones IV-Va, lesiones avanzadas que siguen sin producir sintomatología. En la fase 3, la lesión tipo VI (que son placas complicadas con defectos en la superficie, con hemorragia o depósito de trombo) puede producir clínica de angina o bien, en una fase 4, este mismo tipo de lesión VI, si llega a obstruir por completo el vaso, puede producir síntomas agudos de infarto agudo de miocardio (IAM), angina inestable o muerte súbita. Estas mismas lesiones tipo VI pueden evolucionar hacia la fibrosis o calcificación (lesión tipo Vb-c) y en una fase 5, producir sintomatología de angina o bien constituir una oclusión silente14,17.
En los últimos años se han sistematizado los estudios multicéntricos, y el estudio PBDAY es un ejemplo de ello. Se trata de un estudio llevado a cabo conjuntamente por la OMS y la ISFC (International Society and Federation Cardiology)15 y que recoge casos de autopsias realizadas a población juvenil de cuatro continentes. Para caracterizar la aterosclerosis utiliza las definiciones de lesiones lipídicas, que incluyen depósitos lipídicos que se tiñen con Sudán IV, y lesiones sobreelevadas, áreas de lesión intimal elevadas y duras a la palpación.
los nuevos conocimientos en la patogenia de la aterosclerosis han ido modificando la forma de clasificar la lesión aterosclerótica, y así podemos referirnos básicamente a una primera clasificación, la propuesta por la IAP que ha sido utilizada ampliamente en diversos estudios, y que distingue entre: a) estría grasa o lesiones lipídicas, que consiste en depósitos lipídicos que se tiñen con Sudán IV, todas áreas intimales ligeramente elevadas; b) placa fibrosa o lesiones sobreelevadas (raised lesions) que incluyen áreas de lesión intimal elevadas, duras a la palpación en la inspección macroscópica y que corresponden a placas fibrosas o fibroateromatosas con contenido lipídico o sin él, y c) la lesión calcificada o complicada con hemorragia, ulceración o trombosis.
Una nueva clasificación fue la propuesta en 1992 por Fuster17 basada en la patofisiología y diferenciando tres tipos de lesión. El tipo I supone cambios funcionales en el endotelio con acumulación de monocitos y lípidos en la íntima; el tipo II incluye pérdida de células endoteliales, agregación plaquetaria y proliferación moderada de células musculares lisas, y el tipo III, rotura de lesiones con formación de trombos, importante proliferación de células musculares e incorporación del trombo al interior del vaso causando permanente e importante engrosamiento.
Otra de las clasificaciones, la propuesta y llevada a cabo por Stary, basada en los hallazgos histológicos, distingue entre ocho tipos de lesiones. El American Heart Asociation’s Commitee on Vascular Lesions recomendó hace algunos años una clasificación numérica de los tipos de lesiones definidas histológicamente, y esta clasificación se consideró oportuna y apropiada. Diversos estudios realizados en autopsias utilizando estos métodos histológicos aportaron nueva luz sobre la composición de las lesiones y sobre la diversidad de mecanismos implicados en su desarrollo. Tras la revisión de estos nuevos datos, el Comité recomendó el uso de esta nomenclatura numérica estándar para remplazar a una variedad de términos vagos y duplicados. La clasificación de la AHA se ha desarrollado y usado para comunicar los resultados sobre la composición de lesiones en diversos estudios.
De la clasificación morfológica de Stary puede decirse que es muy útil para tipificar las lesiones ateroscleróticas y que además diferencia el engrosamiento intimal adaptativo de la aterosclerosis inicial, aunque reconoce que en ciertas localizaciones este engrosamiento intimal (del que distingue la forma excéntrica y la difusa) puede favorecer el comienzo de la arteriosclerosis (zonas proclives a la aterosclerosis), mientras que otras zonas son resistentes a no ser que existan concentraciones muy elevadas de lipoproteínas aterogénicas.
En las arterias coronarias normales el grosor de la capa íntima es desigual, encontrándose áreas relativamente delgadas con otras de mayor grosor que representan adaptaciones del vaso en las áreas de mayor flujo sanguíneo y tensión de la pared (engrosamiento adaptativo de la media). Estas áreas están presentes desde el nacimiento, se autolimitan en el crecimiento, no obstruyen el flujo sanguíneo a ninguna edad y generalmente cuando se producen lesiones arterioscleróticas se localizan particularmente en las regiones que muestran dicho engrosamiento adaptativo. En general, el engrosamiento adaptativo de la íntima se encuentra en las regiones próximas a las bifurcaciones arteriales, y en estas áreas el número de macrófagos es hasta tres veces superior al número encontrado en las áreas que no evidencian dicho engrosamiento, y algunos de estos macrófagos pueden contener gotas de lípidos en su citoplasma y constituir lo que denominamos células espumosas1.
Este engrosamiento está compuesto por dos capas distintas, una en contacto con la luz formada por proteoglucanos, escasas fibras elásticas y macrófagos aislados, y otra musculoelástica (capa adyacente a la media) llamada así por su riqueza en células musculares lisas y fibras elásticas, y que también contiene mucho colágeno. El engrosamiento excéntrico es focal y está asociado con ramas y orificios de salida de arterias. El difuso en las coronarias es menor que el excéntrico, aunque más extenso.
Este engrosamiento adaptativo intimal ya fue descrito en aortas fetales humanas por Thoma (1883, 1920). Sin embargo, algunos autores no distinguen este engrosamiento intimal y ha sido designado como aterosclerótico porque es en estos lugares donde comienza la acumulación de lipoproteínas cuando exceden un valor crítico. Como fenómeno adaptativo se acepta que es autolimitado y responde a fuerzas hemodinámicas en localizaciones arteriales específicas.
Clasificación de Stary
Lesión de tipo I
Consiste en cambios iniciales y mínimos que no aumentan el espesor de la pared arterial más allá de lo normal para esta zona. El término lesión inicial también se ha utilizado para definir este tipo de lesión. El sustrato anatómico consiste en pequeños grupos de macrófagos que contienen gotas lipídicas citoplásmicas (macrófagos espumosos) observables en la íntima. Estos cambios son en ocasiones tan sutiles que diferenciar lesiones tipo I de lesiones tipo 0 (normalidad) es una tarea bastante subjetiva, y al interpretar los resultados de numerosos trabajos (teniendo en cuenta que se realizan después del año de vida) ha de suponerse que se parte de lesión tipo I, ya que siempre hay un grado mínimo demacrófagos en la íntima (figs. 4 y 5).
Las lesiones iniciales de aterosclerosis ejemplificadas aquí nos presentan el inicio de la lesión con el cúmulo lipídico en la íntima arterial. Estas lesiones, descritas siguiendo la clasificación de Stary, se presentan uniformemente en todas las poblaciones estudiadas, aunque son más predominantes en las sociedades occidentales con dietas ricas en grasa saturada y colesterol. Ambas lesiones ocurren a edades tempranas, la tipo II corresponde a la llamada estría grasa. Estas lesiones contienen principalmente células espumosas (macrófagos cargados de lípido intracelular). Son lesiones asintomáticas que no estrechan la pared del vaso.
Lesión de tipo II
Incluye lo que macroscópicamente se define como estría grasa, que consiste en placas de coloración amarillenta depositadas en la superficie intimal arterial. Con la técnica de Sudán III o IV estas placas adquieren una tonalidad rojiza, visible en el examen macroscópico, por lo que algunos estudios las han definido como lesiones sudanofílicas. Las características microscópicas de las lesiones tipo II son más evidentes que las tipo I, y contienen mayor número de macrófagos espumosos (es lo que se ha denominado cúmulo intracelular de lípidos). Los estudios microscópicos han determinado que si bien todas las estrías fibrosas corresponden a lesiones tipo II, no todas se manifiestan macroscópicamente en forma de estría grasa (figs. 4 y 5).
Lesión de tipo III
También conocida como lesión intermedia, lesión transicional o preateroma, y se aplica a aquellas lesiones que morfológica y químicamente se encuentran entre las lesiones tipo II y las tipo IV (o ateroma), y que están constituidas por abundantes acumulaciones de macrófagos espumosos, algunos de los cuales vierten este material al exterior, dando lugar a acumulaciones de lípidos extracelulares, en general en escasa proporción (fig. 6).
. La lesión tipo IV (siguiendo la clasificación de Stary) se considera una lesión avanzada. Está formada básicamente por un centro lipídico (flecha) de lípido extracelular en el que en algunos casos pueden encontrarse cristales de colesterol. En su desarrollo y evolución puede sufrir rotura de la capa que cubre la acumulación lipídica y tener lugar una complicación.
Conocida como ateroma, este tipo de lesión es considerada como lesión avanzada debido a la desorganización de la íntima. Se define por la presencia masiva de abundantes acumulaciones de lípidos extracelulares que se observan como masas lipídicas (núcleos lipídicos) al microscopio óptico y suelen localizarse en la capa musculoesquelética. Estas lesiones pueden incluir la presencia de cristales de colesterol, y en algunos casos este centro lipídico es lo bastante grande para observarlo a simple vista cuando se corta la arteria; no obstante, en algunos casos no determinan estrechez del lumen vascular. Las lesiones tipo IV comienzan a aparecer en la segunda mitad de la segunda década de la vida, pueden estrechar la luz arterial sólo mínimamente y no ser visibles angiográficamente, pero ser productoras de síntomas (clínicamente relevantes) por el desarrollo de fisuras en su superficie, hematomas o trombo. Este tipo de lesión suele aparecer en la áreas de engrosamiento intimal adaptativo de tipo excéntrico: luego el ateroma es, al menos inicialmente, una lesión excéntrica. Es el equivalente a lo que otros autores describen como placa, placa fibrolipídica, o placa fibrosa18. Cuando la placa se enriquece en colágeno y después en tejido fibroso, la lesión se etiqueta como tipo V. La importancia potencial del tipo IV puede ser muy importante, aunque no estreche la luz, ya que se considera a este tipo de lesión vulnerable a la rotura debido a la abundancia de macrófagos.
Lesión de tipo V
Las lesiones tipo V, VI, VII y VIII son lesiones ateroscleróticas más avanzadas1,14,15,19,20. La medida biológica usada para designar estas lesiones es la desestructuración y deformidad de una parte de la íntima, que incluye entre otros cambios la presencia de depósitos de
calcio. Se dan desde la cuarta década de la vida. No necesariamente son visibles angiográficamente, y tampoco han de ser lesiones clínicamente manifiestas. Tienen un alto predominio de tejido conjuntivo fibroso; cuando el nuevo tejido es parte de una lesión con un centro lipídico (el tipo IV) este tipo de morfología se denomina fibroateroma o lesión Va. Si presenta zonas de calcificación, se denomina tipo Vb y, por último, si el centro lipídico no existe o es en general mínimo, se llama Vc. Estas lesiones, por regla general, estrechan las arterias más que las tipo IV y desarrollan hendiduras, hematomas y trombosis con importantes consecuencias clínicas.
El aspecto clínico más destacado de estas lesiones consiste en causar el 20% de las muertes coronarias súbitas y los infartos de miocardio que generalmente ocurren en ausencia de trombo luminal. Por otra parte, las lesiones de más del 75% motivan la mayoría de los accidentes vasculares clínicos. Por último, en los pacientes con angina estable tienen una reducción de la luz de más del 50% con una frecuencia de presencia de trombo del 20%. En conclusión, estas lesiones graves estrechan la luz del vaso y presentan una importante reducción del flujo de sangre arterial.
La morbimortalidad de las placas se presenta principalmente en las tipo IV y V, en las que se producen frecuentemente rotura de la superficie de la lesión con hematoma o hemorragia y depósito de trombo19.
Rotura de un aneurisma disecante que recorre toda la longitud de la aorta y produjo la muerte de un sujeto de 67 años. La sangre se introduce entre la íntima y la capa media disecando las dos capas y provocando un robo de sangre del torrente sanguíneo.
Son lesiones complicadas que tienen depósitos trombóticos visibles y hemorragia además de lípido y colágeno (fibroateroma complicado o lesiones complicadas). Se suelen subdividir en VIa: rotura de la superficie con trombo y hemorragia como componentes importantes; tipo VIb: presencia de trombo sin hemorragia, y tipo VIc: hemorragia sin trombo. Clínicamente, las lesiones complicadas con presencia de trombo obstructivo pero lábil se conocen como lesiones inestables, y serían el equivalente morfológico de la angina inestable.
Son múltiples las causas que determinan hemorragia y depósito trombótico. La erosión o ulceración de la lesión es una de las causas bien conocidas. La fisura de la superficie causaría una hemorragia masiva dentro de la lesión, depósito trombótico, rápida expansión de la lesión
y síntomas. Las hemorragias que se producen dentro de la lesión a veces tienen su origen en la rotura de los capilares neoformados. El depósito trombótico en la lesión puede formarse en ausencia de defectos de superficie o hemorragia por cambios en el flujo sanguíneo secundarios a la deformidad que imparte a la superficie una lesión sobreelevada, facilitando el depósito de plaquetas en individuos susceptibles. En personas con episodios isquémicos, se han encontrado niveles de fibrinógeno plasmático elevados.
Muchas lesiones tipo VI aparecen tras la tercera o cuarta década de la vida, tras pasar primero la fase de ateroma (tipo IV); sin embargo, también se han encontrado fisuras o hemorragias masivas asociadas a lesiones de tan sólo estría grasa. En realidad este tipo de lesión es una situación extrema y más avanzada de las lesiones ya descritas de tipo IV y V.
Lesiones tipo VII (lesión calcificada)
En la cuarta década, algunas lesiones aterosclerosas avanzadas se mineralizan, y se les aplica el término lesión tipo VII. El depósito de calcio reemplaza el depósito extracelular de células muertas y lípidos. La cantidad de calcio es variable y, aunque a veces en lesiones tipo IV de jóvenes también pueden verse pequeñas partículas de calcio cristalino con ayuda del microscopio, el tipo VII se usa para describir lesiones con mineralización importante, aunque exista también depósito de lípidos y tejido fibroso.
Lesiones tipo VIII (lesión fibrótica)
Algunas lesiones ateroscleróticas, con frecuencia en arterias de extremidades inferiores, pueden estar formadas enteramente por cicatriz de colágeno, con mínimo componente lipídico o sin él (bien porque haya desaparecido o porque nunca lo haya habido). Pueden obstruir severamente la luz de arterias de mediano calibre u ocluirlas totalmente. El término fibrótico es el que mejor definiría este tipo de lesión.
Formación de trombos
El deterioro funcional o la pérdida de pequeños grupos de células del endotelio vascular, podría facilitar la formación de trombos y evolucionar hacia un defecto de la superficie con hematoma o hemorragia. Esta complicación se relaciona con alteraciones del flujo en las divisiones y acodaduras de las arterias, donde se forman turbulencias21,22.
Las lesiones con trombo pueden sufrir tres procesos diferentes: rotura, erosión y, con menos frecuencia, calcificación del nódulo. La rotura de placa se define como un área de solución de continuidad de una capa fibrosa (a diferencia del trombo que está en continuidad con el
núcleo necrótico). Se da típicamente en grandes núcleos necróticos y en capas fibrosas discontinuas, se encuentra en el 60% de los individuos que mueren súbitamente a causa de un trombo intraluminal y, con menos frecuencia, es causa de muerte en varones de menos de 50 años y mujeres mayores de esa edad. Los factores de riesgo más predictivos para este tipo de lesión son la hipercolesterolemia, escaso colesterol unido a lipoproteínas de alta densidad (cHDL), y elevada razón de colesterol total/cHDL. La erosión de la placa muestra una zona sin endotelio, con exposición de una íntima arterial constituida por células musculares lisas y proteoglucanos y, sorprendentemente, el sitio erosionado contiene mínima inflamación. La erosión de la placa es un fenómeno muy común en mujeres y varones menores de 50 años, constituye el 40% de casos de muertes súbitas cardíacas trombóticas y está asociada al consumo de tabaco, especialmente en mujeres premenopáusicas.
Las lesiones de aterosclerosis avanzadas que contienen trombos son frecuentes en la cuarta década de vida y edades más tardías. En un reciente estudio poblacional con individuos comprendidos entre 30 y 59 años22, se observó que un 38% presentaba trombos en la aorta (figura de trombo ilíaco). Las fisuras y hematomas que se localizan por debajo de los depósitos del trombo en muchos casos pueden repetirse, y esta repetición durante meses o años de hematomas recurrentes y pequeños en una lesión contribuye al estrechamiento gradual de la luz arterial. En algunos casos, el trombo continúa expandiéndose y termina por ocluir el lumen arterial en un lapso relativamente corto (horas o pocos días).
Clasificaciones en controversia
En un reciente artículo sobre muerte súbita de origen cardíaco, Virmany19, después de emitir algunas críticas, lanza una nueva propuesta de clasificación, diferente de la establecida por la AHA. Establece que el esquema de clasificación numérica romana modificada por códigos de letras es una lista muy larga y de difícil recuerdo y que dicha clasificación implica un orden de modelo lineal de progresión de la lesión. Considera que es un sistema ambiguo, porque no deja claro si hay una sola sucesión de eventos durante la progresión de todas las lesiones. Propone un esquema de clasificación más simple y más fácil de utilizar que define siete lesiones: xantoma intimal, engrosamiento intimal, engrosamiento intimal patológico, capa fibrosa de ateroma, capa fibrosa delgada de ateroma, nódulo calcificado y placa fibrocalcificada.
El paradigma actual se basa en la creencia de que la lesión tipo IV (lesión avanzada o ateroma) descrita por la AHA es «estable» porque el núcleo lipídico que constituye el centro de la lesión está cubierto por una consistente capa fibrosa celular. Si aceptamos el argumento de Virchow, esta capa fibrosa que envuelve la masa lipídica (core) de la placa de ateroma, es análoga a la cápsula que contiene un absceso y, al igual que un absceso, la placa puede romperse. La rotura de la capa fibrosa expone el material lipídico «trombogénico», comenzando la agregación plaquetaria, la formación de coágulo y la trombosis y oclusión. La capa fibrosa puede ser gruesa o delgada. Las capas delgadas son las que ser rompen más fácilmente. La cápsula fibrosa delgada se define como aquella con un espesor menor de 65 µm. Como se ha descrito, las lesiones tipo IV y V son las que presentan un centro lipídico y una cápsula fibrosa; pues bien, la rotura de la placa es causante del 60% de las muertes súbitas de origen coronario. Los autores definen la cápsula fibrosa como una capa distinta de tejido conjuntivo que cubre el centro lipídico completamente, formada por células del músculo liso en una matriz de colágeno-proteoglucanos, con grados variables de infiltración de macrófagos y linfocitos.
Progresión de las lesiones y factores implicados
Progresión de la aterosclerosis con la edad
Que el proceso de aterosclerosis se inicia en la infancia es bien conocido desde los años cincuenta, cuando Holman describió, en autopsias de niños norteamericanos de tres o más años de edad, estrías grasas en algunas de las aortas analizadas23.
Las lesiones tipo III pueden aparecer inmediatamente después de la pubertad y por su composición constituyen una transición entre las lesiones tempranas y las avanzadas. Las lesiones tipo IV, frecuentes en la tercera década, son lesiones histológicamente avanzadas. Las lesiones tipo V y VI se inician en la tercera década de la vida y son las lesiones predominantes en personas mayores.
Los resultados obtenidos por McGill et al26, así como los reportados por otros estudios27-30, han mostrado una susceptibilidad especial de la aorta torácica para desarrollar lesiones incipientes tipo estría grasa, pero los estudios anatomopatológicos no encuentran un porcentaje significativo de lesiones avanzadas y no observan por tanto correlación entre la presencia de estría grasa y su progresiva evolución a lesión avanzada, fenómeno este que sí se observa en otros vasos arteriales(coronarias)31.
La extensión de las lesiones en aorta y coronarias, presenta diferencias cuando los grupos de estudio son ajustados por sexo. La estría grasa se desarrolla por igual tanto en aorta como en coronarias de varones y mujeres incluso en la aorta, las mujeres pueden superar al varón en cuanto a número de lesiones24. A mayor edad, las lesiones aórticas son equiparables en ambos sexos, mientras que las lesiones coronarias avanzadas observadas en los varones duplican las observadas en las mujeres32. Diversos estudios de composición química, patrón histológico y con microscopia electrónica evidenciaron que tanto la estría grasa como las lesiones avanzadas en las arterias coronarias y aorta33-38 tienen similitudes en sus componentes estructurales, pero difieren en su cantidad. De ello podríamos concluir que la estría grasa evoluciona a placa fibrosa.
No existe correlación entre la composición y el tamaño de las lesiones con el grado de obstrucción y las manifestaciones clínicas, excepto en el caso de lesiones tipo I-III, las cuales son casi siempre pequeñas y clínicamente silentes. Las lesiones avanzadas del tipo IV al VI pueden obstruir el lumen arterial de arterias de mediano calibre y producir manifestaciones clínicas o, estar presentes y ser clínicamente silentes. Los hallazgos de numerosos estudios anatomopatológicos sugieren que las manifestaciones clínicas están más a menudo asociadas con lesiones menos avanzadas que la lesión tipo VI. Las lesiones tipo IV, por regla general, no obliteran totalmente el lumen arterial, en parte debido a la facilidad de expansión del vaso afectado, pero podrían llegar a obstruirlo totalmente si se incrementan los valores lipídicos con el consecuente aumento de grosor.
Hay pocas dudas o ninguna acerca del papel desempeñado por las placas fibrosas en el desarrollo de coronariopatía. El riesgo se inicia cuando las lesiones avanzadas alcanzan, en sujetos de mediana edad, el 30% de la íntima arterial32. Los individuos con manifestaciones clínicas presentan lesiones avanzadas en aproximadamente el 60% de la superficie intimal27,39,40. Estudios recientes con métodos angiográficos, ultrasonográficos e histoquímicos indican que las características de las lesiones avanzadas son predictivas del riesgo de presentar eventos oclusivos28.
Diversos estudios han servido para explicar el origen de la estría grasa y su evolución progresiva a placa fibrosa. Los macrófagos presentes en la pared arterial poseen receptores con gran afinidad para captar partículas de lipoproteínas de baja densidad (LDL) modificadas por peroxidación lipídica, dando origen a las células espumosas (macrófagos cargados de lípidos). Este proceso puede acelerarse en presencia de grandes concentraciones plasmáticas de colesterol unido a LDL (cLDL) produciéndose una sobrecarga de células espumosas muertas que forman un cúmulo de lípido extracelular. Los macrófagos estimulan la acumulación de lípidos en las células musculares lisas subyacentes, generándose citocinas que a su vez atraen más macrófagos, lo que determina que el proceso inflamatorio se cronifique41. Por lo anteriormente expuesto, podemos deducir que la estría grasa es una manifestación de un proceso fisiológico que puede evolucionar hacia lesiones patológicas bajo ciertas condiciones en las que intervienen mecanismos moleculares y celulares. El origen del depósito lipídico en las placas fibrosas podría ser explicado por un proceso enzimático de hidrólisis de los fosfolípidos de las LDL, mediado por proteoglucanos presentes en la pared arterial. Esta forma de depósito de lípidos extracelulares es distinta de la ya mencionada vía que involucra captación de LDL por células espumosas32,42.
Relación entre los factores de riesgo y progresión de lesión
Las lesiones del endotelio vascular inducidas por factores de riesgo relacionados con el desarrollo de la arteriosclerosis pasan progresivamente de la estría grasa hasta lesiones más avanzadas. No obstante, estudios anatomopatológicos han puesto de manifiesto que cada factor de riesgo ejerce efectos de diversa magnitud según los territorios vasculares estudiados, así por ejemplo, la hipertensión arterial incide selectivamente en la presencia de aterosclerosis de las arterias cerebrales, mientras que el consumo de tabaco incrementa el desarrollo de aterosclerosis en aorta abdominal, arterias ilíacas y femorales43,33. En cada arteria se ha observado que existen áreas sensibles y resistentes al desarrollo de placa45,46, lo que sugiere que los factores de riesgo puedan afectar selectivamente regiones dentro de una arteria en particular, así como territorios vasculares diferentes.
En un pequeño pero significativo estudio cuyos resultados han sido publicados recientemente, se demuestra que en 20 sujetos de edades comprendidas entre los 35 y 86 años la calcificación de arterias coronarias no aumenta la tensión de la cápsula fibrosa ni favorece su rotura; en cambio, las lesiones de aterosclerosis con gran contenido lipídico en su núcleo determinan un espectacular incremento en la tensión de la cápsula fibro-calcificada, favoreciendo su rotura y la complicación de las manifestaciones clínicas48.
El Bogalusa Heart Study49, en sujetos de edades comprendidas entre 6 y 30 años y con lesiones de aterosclerosis aortocoronaria, observó una correlación positiva entre las concentraciones de cLDL, la edad y el porcentaje de superficie de arteria afectada con estría grasa. El PDAY describe la asociación entre índice de masa corporal (IMC) y la mayor extensión de estría grasa y lesión avanzada en la coronaria derecha de los hombres, pero no de la mujeres. Igualmente se han encontrado asociaciones entre tabaco y presencia de lesiones con incremento de células espumosas, y tabaco asociado con mayor extensión de lesiones avanzadas y estría grasa en aorta abdominal, no encontrándose asociación entre tabaco y extensión de lesiones en la coronaria derecha47.
Volviendo a los factores de riesgo lipídicos, este estudio ha puesto de manifiesto que concentraciones bajas de cHDL y altas de colesterol no transportado por HDL están asociadas positivamente con la extensión de la estría grasa y de lesiones avanzadas tanto en aorta abdominal como en coronaria derecha. Por último, la hipertensión favorece sólo el incremento de lesiones avanzadas en ambas arterias. Las observaciones morfológicas indican que existe una progresión continua de estría juvenil a lesión avanzada, que la asociación entre factores de riesgo con estría y lesión avanzada es similar y que la distribución topográfica de ambas lesiones es similar en las coronarias y en la aorta abdominal. Todo ello lleva a concluir que las estrías grasas, bajo ciertas condiciones y en ciertos sitios anatómicos evolucionan a placas fibrosas y eventualmente pueden sufrir otros cambios que directamente causen oclusión. La estría grasa parece ser la lesión inicial del proceso de aterosclerosis, inofensiva si permanece como tal.
Correlación de las lesiones con los síndromes clínicos
Las lesiones de aterosclerosis son el resultado de una variedad de complejos procesos patogénicos, en los que intervienen diferentes factores: formación de células espumosas, acumulación de lípidos extracelulares, desplazamiento, reducción de la matriz intercelular, presencia de células de músculo liso, depósito de calcio, inflamación crónica, neovascularización y, por último, rotura de las lesiones y formación y transformación del hematoma y la trombosis en tejido fibromuscular. Estos procesos son evolutivos, pueden predominar unos sobre otros y dar manifestaciones clínicas.
Síndromes coronarios
Las placas ateromatosas complicadas con procesos de erosión, fisuración o rotura sufren hemorragia, agregación plaquetaria y trombosis. En la mayoría de los casos, todo este proceso cursará asintomático, con un trombo no oclusivo o una hemorragia intraplaca, seguida de una fase de cicatrización, depósito de colágeno y fibrosis, cuyo resultado suele ser una placa estenótica estable. Pero, a veces, si el trombo es oclusivo, se traduce clínicamente como un síndrome coronario agudo.
Estos síndromes se caracterizan por la rapidez de progresión, y la rotura de la placa con el trombo intraluminal resultante desempeña un papel fundamental en la patogenia de los síntomas de insuficiencia coronaria. El grado de rotura de placa y la lesión resultante determinarán los síntomas clínicos. Si la lesión inicial se encuentra sólo en la superficie del endotelio, el estímulo de la trombogénesis es limitado, y lo más frecuente en estos casos es hallar un trombo mural sin síntomas clínicos. Si la lesión subyacente crece y la rotura es más profunda generando una fisura, ocurre el proceso trombótico-oclusivo que puede ser repetitivo en cuestión de minutos, con la consiguiente aparición de síntomas clínicos.
De hecho, algunos pacientes con angina inestable pueden tener oclusión intermitente del vaso afectado e isquemia. El hallazgo anatomopatológico de trombo y fisura es frecuente en los pacientes con infarto o muerte súbita. Por otra parte, la persistencia de lesión y daño celular no hará más que sostener una situación recurrente de trombosis que puede llevar a la oclusión gradual y, por supuesto, a la clínica de angina inestable, infarto o muerte súbita. Finalmente, si se produce la rotura o ulceración con exposición del centro lipídico y de la matriz de colágeno y se desencadenan los mecanismos inherentes a esta rotura, la oclusión trombótica será frecuente y generalmente se traduce clínicamente como infarto agudo de miocardio (entre 2 y 4 h como mucho) (fig. 10).
Infarto agudo de miocardio y rotura miocárdica.
Angina estable crónica y oclusión silenciosa
Los estudios angiográficos realizados en pacientes con angina inestable también se han correlacionado con los hallazgos obtenidos de autopsias. Estas lesiones generalmente tienen un contorno liso, con zonas de adelgazamiento y generalmente simétricas o excéntricas, otras son lesiones ricas en lípidos, generalmente pequeñas, que son propensas a la rotura; por otra parte las lesiones estenóticas severas tienden a ser fibróticas y estables. La presencia de clínica anginosa ocurre cuando el proceso trombótico no es completamente oclusivo, y generalmente es silencioso hasta el momento en que el coágulo se organiza comienza la clínica de angina de esfuerzo.
Las lesiones estenóticas graves tienden a progresar hacia la oclusión, la trombosis y el infarto, aproximadamente tres veces más que las lesiones menos graves, probablemente debido a los vasos colaterales bien desarrollados. En estudios prospectivos angiográficos ha quedado demostrado que los nuevos episodios coronarios se presentan en los pacientes con coronarias estenóticas entre el 35 y el 65%. Esto apoya el concepto de que el potencial trombogénico de las lesiones no requiere de estenosis avanzadas, sino que depende más de las características de la placa subyacente. Por esta razón en los últimos años los esfuerzos se han dirigido a la búsqueda de medidas de estabilización de las placas, fundamentalmente mediante el control de los valores de colesterol, y a la identificación de placas vulnerables con marcadores bioquímicos o mediante técnicas de imagen (resonancia magnética)51 (fig. 3).
El polígono de Willis es uno de los lugares en que existen aneurismas cerebrales. Dichos aneurismas pueden romperse por diversas causas y provocar la muerte del sujeto.
La enfermedad cerebrovascular es la causa de un elevado porcentaje de muertes, invalidez y discapacidad. La aterosclerosis es la causa más común de accidentes cerebrovasculares. Las lesiones anotomopatológicas típicas se encuentran en las bifurcaciones de las arterias carótida interna, vertebral en su origen y cerebral inferior posterior.
Al igual que en la aterosclerosis coronaria, la superficie luminal inestable en las arterias cerebrales es la causa de la lesión, la rotura, la trombosis y el desprendimiento del trombo, lo que determina en última instancia que la enfermedad se manifieste clínicamente. Las roturas, particularmente las localizadas en la bifurcación carotídea, son a menudo una fuente de embolia distal y oclusión, produciendo síntomas cerebrales de isquemia. Sin embargo, lesiones estenóticas importantes de la arteria carótida común e interna, así como de sus ramas intracraneales, pueden llevar a la obstrucción completa, isquemia y necrosis. La presencia de placas en la bifurcación carotídea es causa del 40% de las manifestaciones clínicas, de las que un 20% de las mismas corresponde a las arterias vertebrales.
Trombosis en la bifurcación ilíaca.
El otro territorio vascular con implicaciones clínicas y anatomopatológicas corresponde a la aorta, cuyo segmento abdominal es el que generalmente se encuentra afectado, inmediatamente por debajo de la emergencia de las arterias renales y con afección de la bifurcación ilíaca. Debido al calibre de la aorta, la obstrucción súbita clínicamente significativa es relativamente rara; sin embargo, son frecuentes las lesiones grandes con formación de aneurismas. La aterosclerosis de las arterias ilíacas y femorales es a menudo grave, y en todos los casos predominan los grados morfológicos avanzados, con potencial de causar embolia u obstrucción distal y manifestaciones clínicas típicas de claudicación intermitente.
La aterosclerosis es una de las causas más importantes de formación de aneurismas aórticos. La placas que se originan en la íntima arterial determinan la destrucción de la capa media con adelgazamiento de la pared arterial y formación de la dilatación aneurismática. Los aneurismas ateroscleróticos suelen localizarse en la aorta abdominal, principalmente entre las arterias renales y la bifurcación de las ilíacas, o en las ilíacas comunes, rara vez se desarrollan antes de los 50 años, y son mucho más comunes en varones. Los aneurismas saculares son esencialmente esféricos. La rotura de un aneurisma aórtico puede ser súbita, con las consiguientes hemorragia y muerte. Los aneurismas de crecimiento progresivo y aquellos con dimensiones superiores a 4 cm son potencialmente peligrosos y propensos a la rotura (un riesgo del 4-5% por año)50. Los segmentos de aorta torácico y abdominal son propensos a la formación de placas; sin embargo, por regla general, la presencia de placa y aneurisma en la aorta torácica es menos frecuente y más bien delimitada, y se origina en los orificios intercostales. Effects of lipid-lowering by simvastatin on human atherosclerotic lesions: a longitudinal study by high-resolution, noninvasive magnetic resonance imaging..
. Un nuevo enfoque en el tratamiento de las placas de ateroma
Con la eliminación de un determinado gen en las células endoteliales se consigue reducir de forma significativa tanto la inflamación como la placa en los vasos sanguíneos
El Médico Interactivo
5 de septiembre 2019. 1:43 pm
En un estudio realizado en la Universidad de Yale, los investigadores han revelado factores previamente desconocidos que contribuyen al endurecimiento de las arterias y el crecimiento de la placa, lo que puede ser la base de un enfoque prometedor para detener la acumulación de placa.
Los tratamientos actuales sirven para retrasar la enfermedad pero no para mejorarla, lo cual puede deberse a la inflamación continua en los vasos sanguíneos, según los expertos. Para comprender los factores que contribuyen a esta inflamación, los investigadores se centraron en un grupo de proteínas llamado factor de crecimiento transformante beta (TGFB), que regula una amplia gama de células y tejidos en todo el organismo.
Utilizando células humanas cultivadas, los investigadores descubrieron que las proteínas TGFB provocan inflamación en las células endoteliales, pero no en otros tipos de células.
A través de una técnica que mide la expresión de cada gen en células individuales, mostraron que el TGFB inducía una inflamación en estas células. Los resultados se publican en la revista Nature Metabolism.
“Este hallazgo es notable han destacado los investigadores, porque se sabe que las proteínas TGFB disminuyen la inflamación en otras células del organismo”.
Los autores también mostraron que cuando se elimina el gen del receptor de TGFB en las células endoteliales, tanto la inflamación como la placa en los vasos sanguíneos se reducen significativamente.
Para probar la terapia, el equipo utilizó ARNi, un fármaco desarrollado en Yale que interrumpe los receptores de TGFB. Este ARN interferente usa la propia secuencia de ADN de un gen para apagar o silenciar el gen. Aplicaron el medicamento sólo a las células endoteliales de las paredes de los vasos sanguíneos. Esta estrategia redujo la inflamación y la placa tan efectivamente como la técnica genética.
“Los hallazgos, aseguran, identifican la señalización de TGFB como una causa importante de inflamación crónica de la pared de los vasos, y demuestran que la interrupción de esta vía conduce al cese de la inflamación y a una regresión sustancial de la placa existente”, concluyen. La placa aterogénica: fisiopatología y consecuencias clínicas
A. Bertomeu Ruiza, D. Zambón Radosa
a Secci??n de L??pidos. Servicio de Diet??tica y Nutrici??n. Hospital Cl??nic. Barcelona.
Este artículo ha recibido
43012
 William Beecher Scoville, cirujano que intervino a Henry Molaison
William Beecher Scoville, cirujano que intervino a Henry Molaison


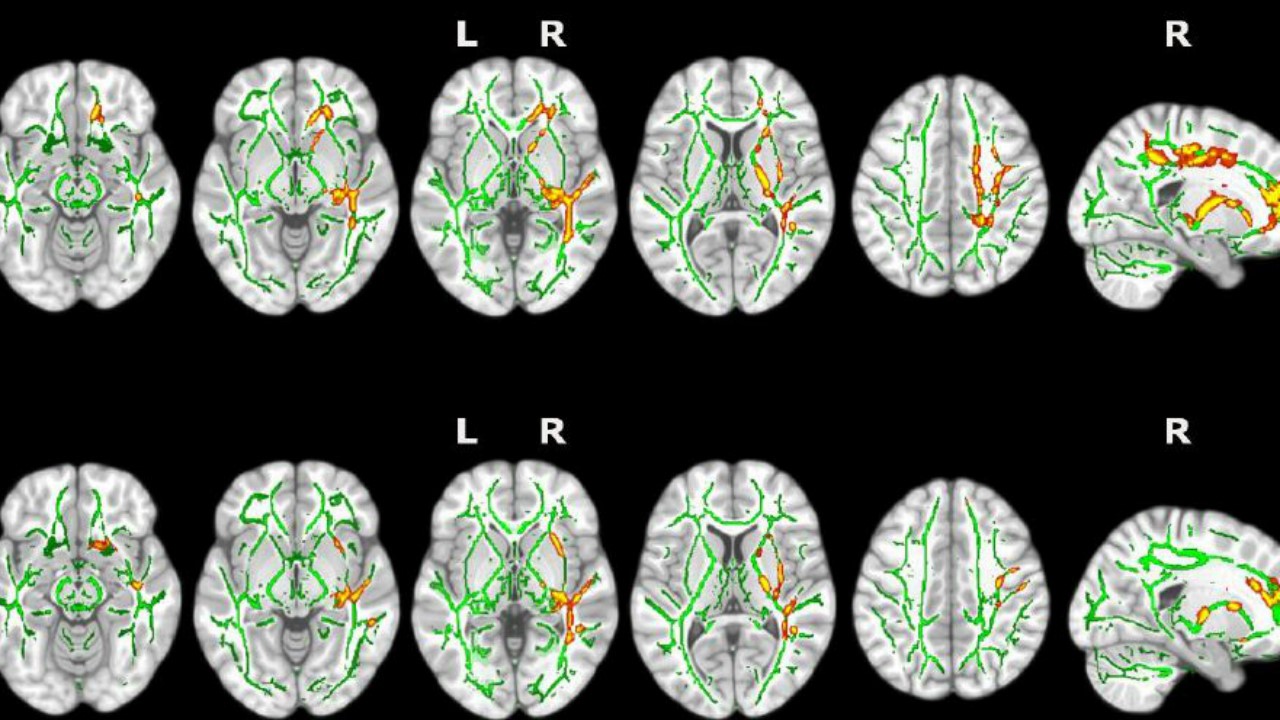 EL INSOMNIO CAUSA ALTERACIONES CEREBRALES PRECURSORAS DE ALZHEIMER
EL INSOMNIO CAUSA ALTERACIONES CEREBRALES PRECURSORAS DE ALZHEIMER />ENTENDER EL CEREBRO
/>ENTENDER EL CEREBRO
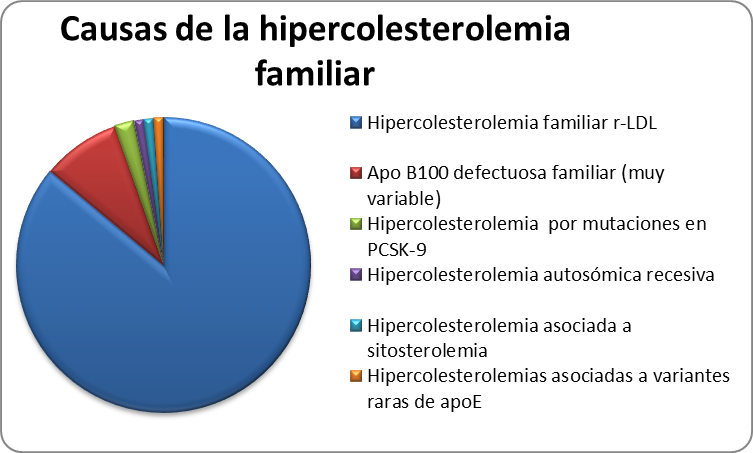
 Por su mecanismo de transmisión, se reconocen dos variantes:
Por su mecanismo de transmisión, se reconocen dos variantes: