LA NEUROFIBROMATOSIS)
La neurofibromatosis es un síndrome neurocutáneo
La neurofibromatosis se refiere a varios trastornos relacionados que tienen manifestaciones clínicas que se solapan, que tienen causas genéticas distintas. Esto causa varios tipos de tumores benignos o malignos que afectan nervios centrales o periféricos y a menudo causa máculas pigmentadas de la piel y, a veces otras manifestaciones. El diagnóstico es clínico. No existe un tratamiento específico, pero los tumores benignos se puede extirpar quirúrgicamente, y los tumores malignos (que son menos comunes) puede ser tratados con radioterapia o quimioterapia.
Hay 3 tipos principales de neurofibromatosis (NF): NF1, NF2 y schwannomatosis, que son causadas por mutaciones de genes.
La NF1 provoca en forma típica alteraciones cutáneas, neurológicas y óseas, pero puede afectar casi cualquier parte del cuerpo.
NF2 produce neurinomas bilaterales del acústico.
La schwannomatosis provoca múltiples schwannomas no intradérmicos; no causa neurinomas del acústico.
El diagnóstico se realiza mediante criterios clínicos; se realizan neuroimágenes si los pacientes tienen anomalías neurológicas.
No existe un tratamiento específico, pero los neurofibromas que causan síntomas graves se puede extirpar quirúrgicamente.
Los tumores malignos pueden requerir quimioterapia.
La neurofibromatosis tipo 1 (NF1, o enfermedad de von Recklinghausen) es más frecuente y ocurre en 1 de 2.500 a 3.000 personas. Esta enfermedad causa manifestaciones neurológicas, cutáneas y, a veces, óseas o de partes blandas. El gen de la NF1 se encuentra en la banda 17q11.2 y codifica la síntesis de neurofibromina; se han identificado > 1.000 mutaciones. Aunque es un trastorno autosómico dominante, 20 a 50% de los casos son causados por una mutación de células germinales de novo.
La neurofibromatosis tipo 2 (NF2) representa el 10% de los casos y se presenta en aproximadamente 1 de 35.000 personas. Se manifiesta principalmente como neuromas acústicos bilaterales congénitos (schwannomas vestibulares). El gen de la NF2 se encuentra en la banda 22q11 y codifica la síntesis de merlina, un supresor de tumores; se han identificado 200 mutaciones. La mayoría de las personas con NF2 lo han heredado de uno de sus padres.
La schwannomatosis, un trastorno poco común, se clasifica como tercer tipo de neurofibromatosis. En 15% de los casos, este tipo es familiar y está relacionado con una mutación germinal en el gen SMARCB1, un gen supresor tumoral localizado en el cromosoma 22q11.23, muy cerca del gen NF2. En los casos restantes, no se conoce bien la base genética, pero en el tejido de algunos pacientes, otras mutaciones del mismo gen están involucradas. Dos o más schwannomas se desarrollan en los nervios vertebrales y en los nervios periféricos y son a veces muy dolorosos; sin embargo, no se desarrollan los neuromas acústicos. La schwannomatosis solía ser considerada una forma de NF2 porque hay múltiples schwannomas en ambas condiciones; sin embargo, el cuadro clínico es diferente, y los genes implicados son distintos.
Neurofibromas periféricos y centrales
Los tumores pueden ser periféricos o centrales.
Los tumores periféricos son frecuentes en la NF1 y pueden aparecer en cualquier lugar del recorrido de nervios periféricos. Los tumores son neurofibromas, que se desarrollan a partir de las vainas nerviosas y consisten en mezclas de células de Schwann, fibroblastos, células neurales y mastocitos. La mayoría aparece durante la adolescencia. De vez en cuando, se transforman en tumores malignos de la vaina nerviosa. Hay múltiples formas:
Los neurofibromas cutáneos son blandos y carnosos.
Los neurofibromas subcutáneos son duros y nodulares.
Los neurofibromas plexiformes nodulares pueden afectar las raíces de los nervios espinales y, en general, crecen hacia el agujero intervertebral para dar origen a masas intraespinales y extraespinales (tumor en reloj de arena). La parte intraespinal puede comprimir la médula espinal.
Los neurofibromas plexiformes difusos (nódulos subcutáneos o crecimiento amorfo excesivo del hueso o las células de Schwann subyacentes) pueden causar desfiguración y déficits distales al neurofibroma. Los neurofibromas plexiformes pueden malignizar y parecen ser los precursores más comunes de los tumores malignos de la vaina del nervio periférico en personas con NF1.
Los schwannomas derivan de las células de Schwann, rara vez sufren transformación maligna y pueden ocurrir en los nervios periféricos en cualquier parte del cuerpo.
Los tumores centrales tienen varias formas:
Gliomas ópticos: estos tumores son astrocitomas pilocíticos de bajo grado, que pueden ser asintomáticos o pueden progresar lo suficiente para comprimir el nervio óptico y causar ceguera. Se producen en los niños más pequeños; estos tumores pueden por lo general ser identificados hacia los 5 años y rara vez se desarrollan después de los 10 años. Aparecen en la NF1.
Neurinomas del acústico (schwannomas vestibulares): estos tumores pueden causar mareos, ataxia, sordera y acúfenos debido a la compresión del nervio craneal VIII; a veces causan debilidad facial debido a la compresión de la VII nervio adyacente. Ellos son el rasgo distintivo de la NF2.
Meningiomas: estos tumores se desarrollan en algunas personas, particularmente aquellos con NF2.
Signos y síntomas de la neurofibromatosis
Neurofibromatosis tipo 1 (NF1)
La mayoría de los pacientes con NF-1 son asintomáticos. Algunos presentan síntomas neurológicos o deformidades óseas. En > 90%, las lesiones cutáneas características son evidentes en el momento del nacimiento o aparecen durante la lactancia.
Las lesiones de café con leche son máculas similares a pecas de color café con leche, distribuidas la mayoría de las veces sobre el tronco, la pelvis y los pliegues flexores de codos y rodillas. Aunque los niños que no tienen neurofibromatosis pueden tener 2 o 3 manchas café con leche, los niños con NF1 tienen ≥ 6 de tales máculas y, a menudo muchas más. Estas máculas son > 5 mm en los niños prepúberes afectados y > 15 mm en los pacientes pospúberes (véase tabla Diagnóstico de la neurofibromatosis).
Son frecuentes los neurofibromas cutáneos que surgen a lo largo de pequeños nervios periféricos. Durante etapas tardías de la infancia, aparecen estos tumores cutáneos de color carne y tamaños y formas variables, que varían en número de varios a miles. Suelen ser asintomáticas.
Los neurofibromas plexiformes pueden desarrollarse y tienden a crecer hasta alcanzar grandes tamaños, lo que produce un engrosamiento irregular y distorsión de las estructuras, que a veces presentan deformidades grotescas responsables de la compresión de los nervios y otras estructuras. Los neurofibromas plexiformes también pueden involucrar a los nervios craneales, típicamente el V, el IX y el X.
Los síntomas neurológicos varían según la localización y el número de neurofibromas. Los neurofibromas más grandes pueden comprimir su nervio de origen y causar parestesias distales, dolor e hipoestesia o debilidad, dependiendo de la función del nervio que está involucrado. Los neurofibromas que se forman a lo largo de las raíces nerviosas espinales, especialmente donde las raíces nerviosas están contenidas por el hueso, pueden comprimir las raíces nerviosas y causar dolor radicular, debilidad o pérdida generalizada de la sensibilidad en esa distribución nerviosa. Los neurofibromas plexiformes que comprimen los nervios craneales causan déficits típicos de esos nervios.
Manifestaciones cutáneas de la neurofibromatosis tipo 1
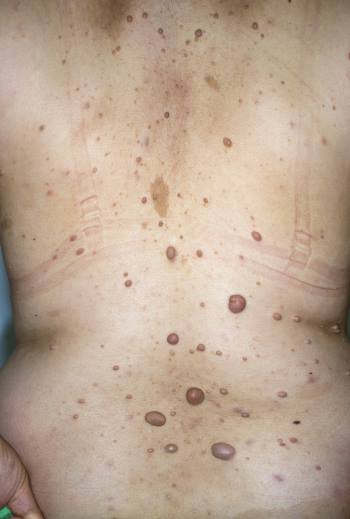
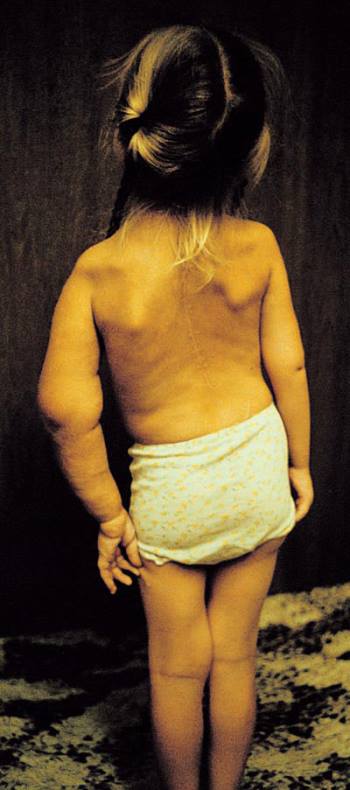
Neurofibromas y manchas de café con leche (espalda)
Esta fotografía muestra múltiples neurofibromas (nódulos pardos en relieve) y manchas «café con leche» (manchas planas pardas) en la espalda de un paciente con neurofibromatosis.
DR HAROUT TANIELIAN/SCIENCE PHOTO LIBRARY
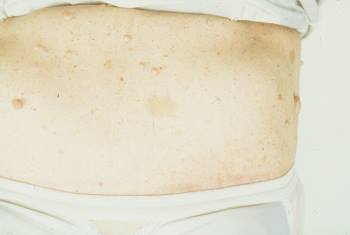
Neurofibromas y manchas de café con leche
Esta fotografía muestra neurofibromas (nódulos pardos en relieve) y manchas «café con leche» (manchas planas pardas) en un paciente con neurofibromatosis.
Imagen cortesía de Karen McKoy, MD.
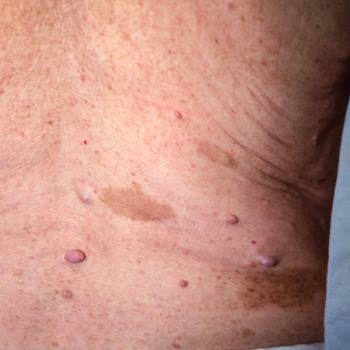
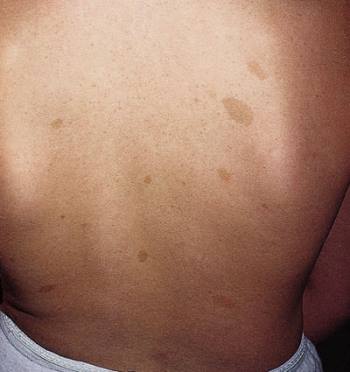
Neurofibromatosis (máculas café con leche y neurofibromas)
Este paciente con neurofibromatosis tipo 1 tiene varias manchas de color café con leche (máculas hiperpigmentadas) y neurofibromas (nódulos levantados).
© Springer Science+Business Media
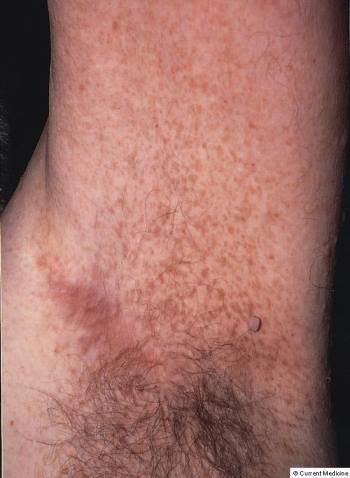
Manifestaciones cutáneas en la neurofibromatosis tipo 1 (NF1)
Esta fotografía muestra máculas axilares semejantes a pecas en la NF1.
© Springer Science+Business Media
Neurofibromatosis (máculas café con leche)
Estas lesiones son máculas con bordes netos presentes en la mayoría de los pacientes con neurofibromatosis de tipo 1 (NF1).
From Oster A, Rosa R: Atlas of Ophthalmology. Edited by R Parrish II and HW Flynn Jr. Philadelphia, Current Medicine, 2000.
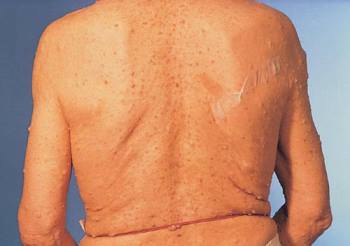
Neurofibromatosis (Neurofibromas) La espalda y los brazos de este hombre de 69 años están cubiertos de pequeños neurofibromas, y hay una mancha café con leche justo medial a su escápula derecha.
Las alteraciones óseas son
Quiste óseos subperiósticos
Festoneado vertebral
Adelgazamiento de la cortical de los huesos largos
Seudoartrosis
Ausencia del ala mayor del hueso esfenoides (pared posterior de la órbita), con el consiguiente exoftalmos pulsátil
Neurofibromatosis (anomalías esqueléticas)
Se observa un neuroma plexiforme en el brazo izquierdo de la paciente, que se extiende desde el húmero proximal hasta la mano. El húmero presenta múltiples fracturas diafisarias asociadas con adelgazamiento de la corteza ósea y seudoartrosis. La paciente también presenta escoliosis, baja talla y agrandamiento del conducto intramedular lumbar causado por meningocele anterior.
En algunos pacientes, se observan glioma óptico y nódulos de Lisch (hamartomas del iris). Los gliomas ópticos suelen ser asintomáticos y no requieren tratamiento a menos que aumenten progresivamente de tamaño.
Los pacientes con NF1 también pueden tener un síndrome de Moyamoya (estenosis u oclusión de las arterias en y alrededor del círculo de Willis con formación de pequeñas arterias colaterales), o aneurismas de la arteria intracraneal. Algunos niños tienen problemas de aprendizaje y ligera macrocefalia.
Neurofibromatosis (nódulos de Lisch)
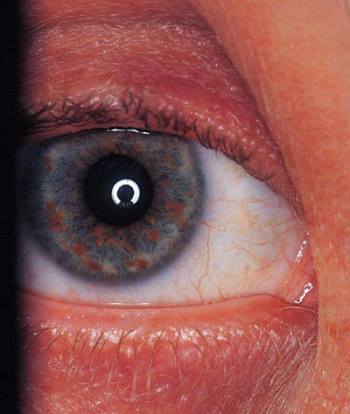
Ocultar los detalles
Los nódulos de Lisch en el iris son hamartomas melanóticos hiperpigmentados.
.
Los niños y adolescentes con NF1 pueden tener leucemia mielomonocítica crónica infantil (leucemia mielomonocítica juvenil) y rabdomiosarcoma. Los feocromocitomas pueden manifestarse a cualquier edad.
Los tumores malignos son mucho menos comunes, pero aún siguen siendo más frecuentes que en la población general; ellos incluyen los gliomas supratentoriales o del tronco cerebral y la transformación de neurofibromas plexiformes a los tumores malignos de la vaina nerviosa. Estos tumores pueden desarrollarse a cualquier edad.
Neurofibromatosis tipo 2 (NF2)
En la NF2 se desarrollan neuromas acústicos bilaterales y se tornan sintomáticos durante la infancia o la edad adulta temprana. Causan hipoacusia, inestabilidad y, a veces, cefalea o debilidad facial. Puede haber masas bilaterales en el octavo par craneal (nervio vestibulococlear). Los miembros de la familia pueden tener gliomas, meningiomas o schwannomas.
Schwannomatosis
En la schwanomatosis, múltiples schwannomas se desarrollan en los nervios craneales, espinales y periféricos. No se desarrollan neurinomas del acústico y los pacientes no se vuelven sordos. Además, los otros tipos de tumores que a veces se producen en los trastornos neurocutáneos no se desarrollan.
El primer síntoma de la schwanomatosis suele ser el dolor, que puede llegar a ser crónico e intenso. Se pueden desarrollar otros síntomas, lo que depende de la localización de los schwannomas.
Evaluación clínica
RM cerebral o TC de cráneo
La mayoría de los pacientes con NF1 se identifican durante exámenes sistemáticos, exámenes por consultas estéticas o evaluaciones de antecedentes familiares positivos. (Véase también guidelines for the diagnosis and management of individuals with NF1 del Consejo Asesor Clínico de la Neurofibromatosis Association del Reino Unido, guidelines for the health supervision of children with NF1 de una colaboración de expertos, clinical practice guidelines for the care of adults with NF1 del American College of Medical Genetics and Genomics.)
El diagnóstico de los 3 tipos es clínico ( ver Diagnóstico de neurofibromatosis) mediante una exploración física detallada centrada en los sistemas cutáneo, esquelético y neurológico. Debe sospecharse y vigilarse la presencia de NF1 en niños que tienen múltiples manchas café con leche, incluso si no tienen otras características o antecedentes familiares de NF1.
La RM cerebral se realiza en pacientes con síntomas o signos neurológicos y, cuando no es posible una evaluación visual detallada, en los niños más pequeños que cumplen los criterios clínicos para la NF1 y que pueden tener un glioma óptico. La RM ponderada en T2 puede mostrar engrosamiento o tortuosidad de los nervios ópticos y lesiones hiperintensas del parénquima que cambian con el tiempo y se correlacionan con pequeñas estructuras quísticas en NF1; la RM puede ayudar a identificar neuromas acústicos o meningiomas en la NF2. Si se sospecha un neuroma acústico, se puede realizar una TC de la cresta petrosa; que por lo general muestra ensanchamiento del conducto auditivo.
Las pruebas genéticas no se realizan normalmente en estos trastornos porque no todas las mutaciones son conocidas y los criterios clínicos son claros.
Tratamiento de la neurofibromatosis
Para los neurofibromas sintomáticos, posiblemente cirugía o extirpación con láser o electrocauterio
Para los tumores malignos, quimioterapia
No se dispone de un tratamiento general para la neurofibromatosis.
Los neurofibromas que causan síntomas graves pueden requerir resección quirúrgica o, si son pequeños, eliminación con láser o electrocauterio. La extirpación quirúrgica de los neurofibromas plexiformes puede anular la función del nervio afectado, y los neurofibromas tienen una tendencia a recurrir en el sitio de la extirpación. En la actualidad se llevan a cabo ensayos clínicos sobre varios tratamientos médicos para los neurofibromas plexiformes y espinales, como por ejemplo con el uso de Sirolimús.
La mayoría de los gliomas ópticos son asintomáticos y solo es necesario controlarlos para determinar su progresión. Tanto para los gliomas ópticos progresivos como para las lesiones del sistema nervioso central que se han vuelto malignas, la quimioterapia es el tratamiento de elección.
Si alguno de los progenitores tiene una neurofibromatosis, el riesgo para la descendencia ulterior es del 50%; si ninguno de los progenitores presenta la enfermedad, el riesgo de los hijos siguientes es incierto, porque son frecuentes las mutaciones nuevas, sobre todo en NF1.
Referencias
From Kotagal S, Bicknese A, Eswara M: Atlas of Clinical Neurology. Edited by RN Rosenberg. Philadelphia, Current Medicine, 2002.
From Bird T, Sumi S: Atlas of Clinical Neurology. Edited by RN Rosenberg. Philadelphia, Current Medicine, 2002
HAROUT TANIELIAN/SCIENCE PHOTO LIBRARY
From Kotagal S, Bicknese A, Eswara M: Atlas of Clinical Neurology. Edited by RN Rosenberg. Philadelphia, Current Medicine, 2002